After getting through the initial MAKER annotation and SNAP gene predictions, I needed (wanted?) to simplify annotation names that will be easier to read and are in a more standardized format; similiar to NCBI.
MAKER provides this functionality.
Ran the following SBATCH script on Mox:
#!/bin/bash
## Job Name
#SBATCH --job-name=maker
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=15-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/outputs/20190108_oly_maker_id_mapping
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Load Open MPI module for parallel, multi-node processing
module load icc_19-ompi_3.1.2
# SegFault fix?
export THREADS_DAEMON_MODEL=1
# Document programs in PATH (primarily for program version ID)
date >> system_path.log
echo "" >> system_path.log
echo "System PATH for $SLURM_JOB_ID" >> system_path.log
echo "" >> system_path.log
printf "%0.s-" {1..10} >> system_path.log
echo ${PATH} | tr : \\n >> system_path.log
# Variables
maker_dir=/gscratch/srlab/programs/maker-2.31.10/bin
maker_prot_fasta=/gscratch/scrubbed/samwhite/outputs/20181127_oly_maker_genome_annotation/snap02/20181127_oly_genome_snap02.all.maker.proteins.fasta
maker_transcripts_fasta=/gscratch/scrubbed/samwhite/outputs/20181127_oly_maker_genome_annotation/snap02/20181127_oly_genome_snap02.all.maker.transcripts.fasta
snap02_gff=/gscratch/scrubbed/samwhite/outputs/20181127_oly_maker_genome_annotation/snap02/20181127_oly_genome_snap02.all.gff
cp ${maker_prot_fasta} 20181127_oly_genome_snap02.all.maker.proteins.renamed.fasta
cp ${maker_transcripts_fasta} 20181127_oly_genome_snap02.all.maker.transcripts.renamed.fasta
cp ${snap02_gff} 20181127_oly_genome_snap02.all.renamed.gff
# Run MAKER programs
## Change gene names
${maker_dir}/maker_map_ids \
--prefix OLUR_ \
--justify 8 \
${snap02_gff} \
> 20181127_oly_genome.map
## Map GFF IDs
${maker_dir}/map_gff_ids \
20181127_oly_genome.map \
20181127_oly_genome_snap02.all.renamed.gff
## Map FastAs
### Proteins
${maker_dir}/map_fasta_ids \
20181127_oly_genome.map \
20181127_oly_genome_snap02.all.maker.proteins.renamed.fasta
### Transcripts
${maker_dir}/map_fasta_ids \
20181127_oly_genome.map \
20181127_oly_genome_snap02.all.maker.transcripts.renamed.fasta
RESULTS
Output folder:
Mapping file (text):
Renamed GFF:
Renamed protein FastA:
Renamed transcripts FastA:
Here’s a look at the files to show that this worked.
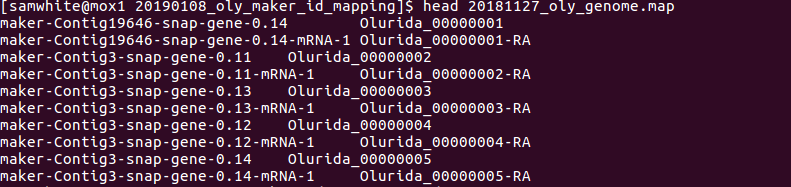
- Map file
This files is used as a key for the name conversions, original names on left, new names on right
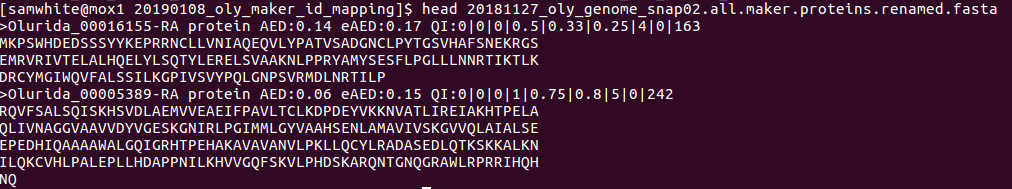
- Proteins FastA
Note the name update at the beginning of the FastA descriptions: Olurida_#########-RA
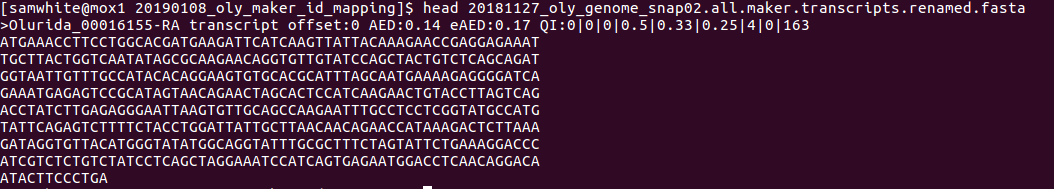
- Transcripts FastA
Note the name update at the beginning of the FastA descriptions: Olurida_#########-RA
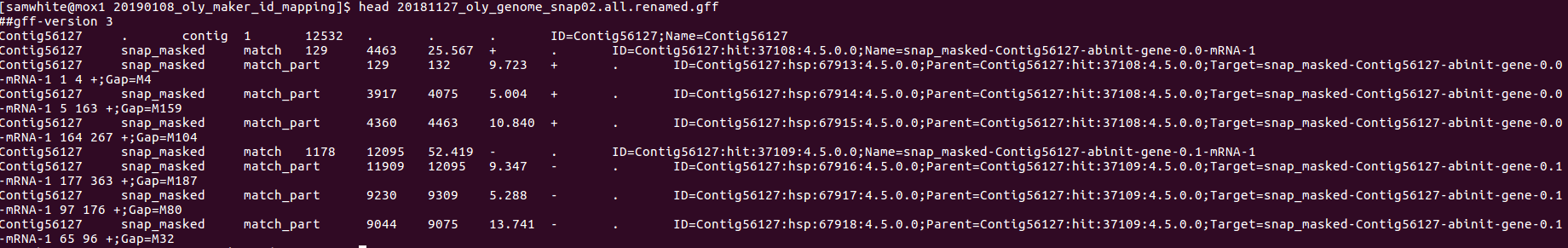
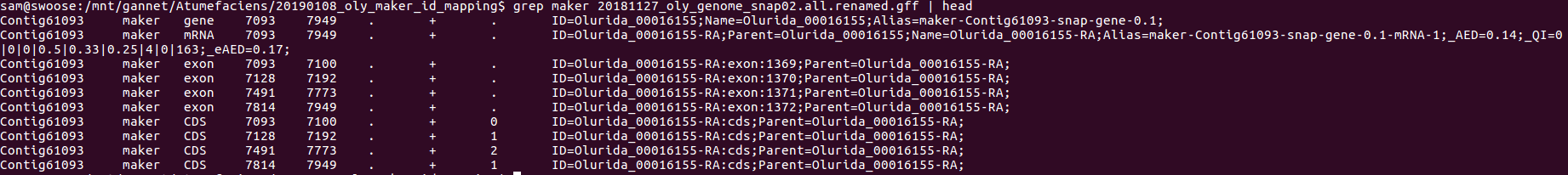
- GFF
The new IDs are present but only shown for gene models:
Regardless, the next steps are to use the newly labeled protein FastA file for BLASTp and protein domain identification. Will move forward with those two steps.