UPDATE 20191125
Since the results I obtained on my final attempt to get this to work failed, I decided to double-check the primer sequences.
Well, I ordered/used the wrong sequences! The two general Crassostrea spp. primers ordered were the 28s primers listed in that paper, instead of the cytochrome oxidase primers! I’ve ordered the correct universal CO primers [which are actually listed in this paper: (Folmer, O., M. Black, W. Hoeh, R. Lutz & R. Vrijenhoek. 1994. DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrate. Mol. Mar. Biol. Biotechnol. 3:294–299.) and will re-run this.
I’m leaving the original post below for posterity:
After isolating DNA earlier today, I ran PCRs on all the samples.
Primers and cycling parameters were taken from this publication:
| SR ID | Primer Name | Sequence |
|---|---|---|
| 1727 | COreverse | CAGGGGGCCGTTCGCGGTCAACGCT |
| 1726 | COCsi546r | AAGTAACCTTAATAGATCAGGGAACC |
| 1725 | COCgi269r | TCGAGGAAATTGCATGTCTGCTACAA |
| 1724 | COforward | GGGACTACCCCCTGAATTTAAGCAT |
This is a multiplex PCR, where the COforward/reverse primers should amplify any Crassostrea spp. DNA (i.e. a positive control - 697bp) and the other two primers will amplify either C.gigas (Cgi269r - 269bp) or C.sikamea (Csi546r - 546bp).
I ended up running this PCR two times, due to:
Ran the gel too far and ran most of the target products off the gel!
Many non-specific bands produced.
First PCR info:
| Component | Single Rxn Vol. (uL) | Num. Rxns | Total Volumes (uL) |
|---|---|---|---|
| DNA | 4 | NA | NA |
| 2x Apex Red Master Mix | 12.5 | 18 | 225 |
| P1 Mix | 1.5 | 18 | 27 |
| P2 Mix | 1.5 | 18 | 27 |
| H2O | 5.5 | 18 | 99 |
| 25 | Add 21uL to each PCR tube |
Cycling params:
95oC for 10mins
30 cycles of:
- 95oC 1min
- 51oC 1min
- 72oC 1min
72oC 10mins
Run on 0.8% agarose gel.
Since the first PCR didn’t work, I made some changes:
Longer gel (1.5% agarose) to improve ability to resolve lower molecular weight bands.
Increase annealing temp and reduce extension time to try to increasae specificity.
Add primers individually instead of primer mixes. Used 100uM stocks.
| Component | Single Rxn Vol. (uL) | Num. Rxns | Total Volumes (uL) |
|---|---|---|---|
| DNA | 4 | NA | NA |
| 2x Apex Red Master Mix | 12.5 | 18 | 225 |
| COforward | 0.15 | 18 | 2.7 |
| Coreverse | 0.15 | 18 | 2.7 |
| COCgi269r | 0.1 | 18 | 1.8 |
| COCsi546r | 0.1 | 18 | 1.8 |
| H2O | 8 | 18 | 144 |
| 25 | Add 21uL to each PCR tube |
Cycling params:
95oC for 10mins
30 cycles of:
- 95oC 1min
- 55oC 1min
- 72oC 45secs
72oC 10mins
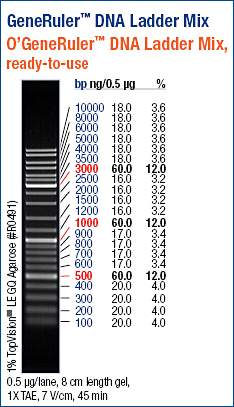
Used the GeneRuler DNA Ladder Mix (ThermoFisher) for all gels:
RESULTS
Gel #1

Overall, this gel is a mess, so I’m haven’t bothered taking the time to label anything. The ladders should be obvious. The no template control (NTC) is in the lane on the furthest right side of the gel and is blank, as it should be.
All other lanes show many bands, most of them larger than the expected largest band size (697bp).
In addition to that, I ended up running the gel too long (in an attempt to get better resolution of the lower molecular weight PCR products), so I lost any bands <500bp.
Gel #2
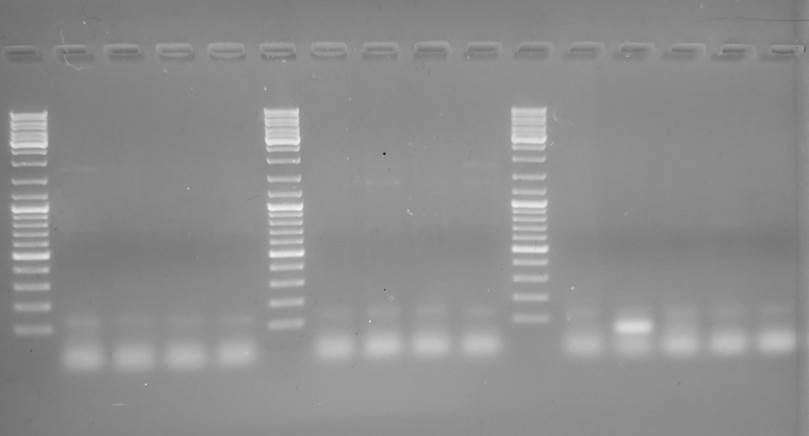
Although this gel is cleaner and has the full range of the ladder and encompasses all potential band sizes, there’s no amplification in any lane; just primer dimers. That suggests that the changes I made to the cycling parameters were no good.
Guess I’ll repeat the PCR using the original cycling parameters and I’ll be sure to run the products on a large gel to get a better idea of what’s happening here.