I previously annotated reads and converted them to the MEGAN6 format RMA6 on 20200318.
I’ll use the MEGAN6 GUI to “Open” the RMA6 file. Once the file loads, you get a nice looking taxonomic tree! From here, you can select any part of the taxonomic tree by right-clicking on the desired taxonomy and “Extract reads…”. Here, you have the option to include “Summarized reads”. This option allows you to extract just the reads that are part of the exact classification you’ve selected or all those within and “below” the classification you’ve selected (i.e. summarized reads).
Extracted reads will be generated as FastA files.
Example:
If you select Arthropoda and do not check the box for “Summarized Reads” you will only get reads classified as Arthropoda! You will not get any reads with more specific taxonomies. However, if you select Arthropoda and you do check the box for “Summarized Reads”, you will get all reads classified as Arthropoda AND all reads in more specific taxonomic classifications, down to the species level.
I will extract reads from two phyla:
Arthropoda (for crabs)
Alveolata (for Hematodinium)
After read extractions using MEGAN6, I’ll need to extract the actual reads from the trimmed FastQ files. This will actually entail extracting all trimmed reads from two different sets of RNAseq:
It’s a bit convoluted, but I realized that the FastA headers were incomplete and did not distinguish between paired reads. Here’s an example:
R1 FastQ header:
@A00147:37:HG2WLDMXX:1:1101:5303:1000 1:N:0:AGGCGAAG+AGGCGAAG
R2 FastQ header:
@A00147:37:HG2WLDMXX:1:1101:5303:1000 2:N:0:AGGCGAAG+AGGCGAAG
However, the reads extracted via MEGAN have FastA headers like this:
>A00147:37:HG2WLDMXX:1:1101:5303:1000
SEQUENCE1
>A00147:37:HG2WLDMXX:1:1101:5303:1000
SEQUENCE2Those are a set of paired reads, but there’s no way to distinguish between R1/R2. This may not be an issue, but I’m not sure how downstream programs (i.e. Trinity) will handle duplicate FastA IDs as inputs. To avoid any headaches, I’ve decided to parse out the corresponding FastQ reads which have the full header info.
Here’s a brief rundown of the approach:
Create list of unique read headers from MEGAN6 FastA files.
Use list with
seqtkprogram to pull out corresponding FastQ reads from the trimmed FastQ R1 and R2 files.
This aspect of read extractions/concatenations is documented in the following Jupyter notebook (GitHub):
RESULTS
OUTPUT FOLDERS
Initial reads extracted as FastAs:
FastQ C.bairdi read extractions:
FastQ Hematodinium read extractions:
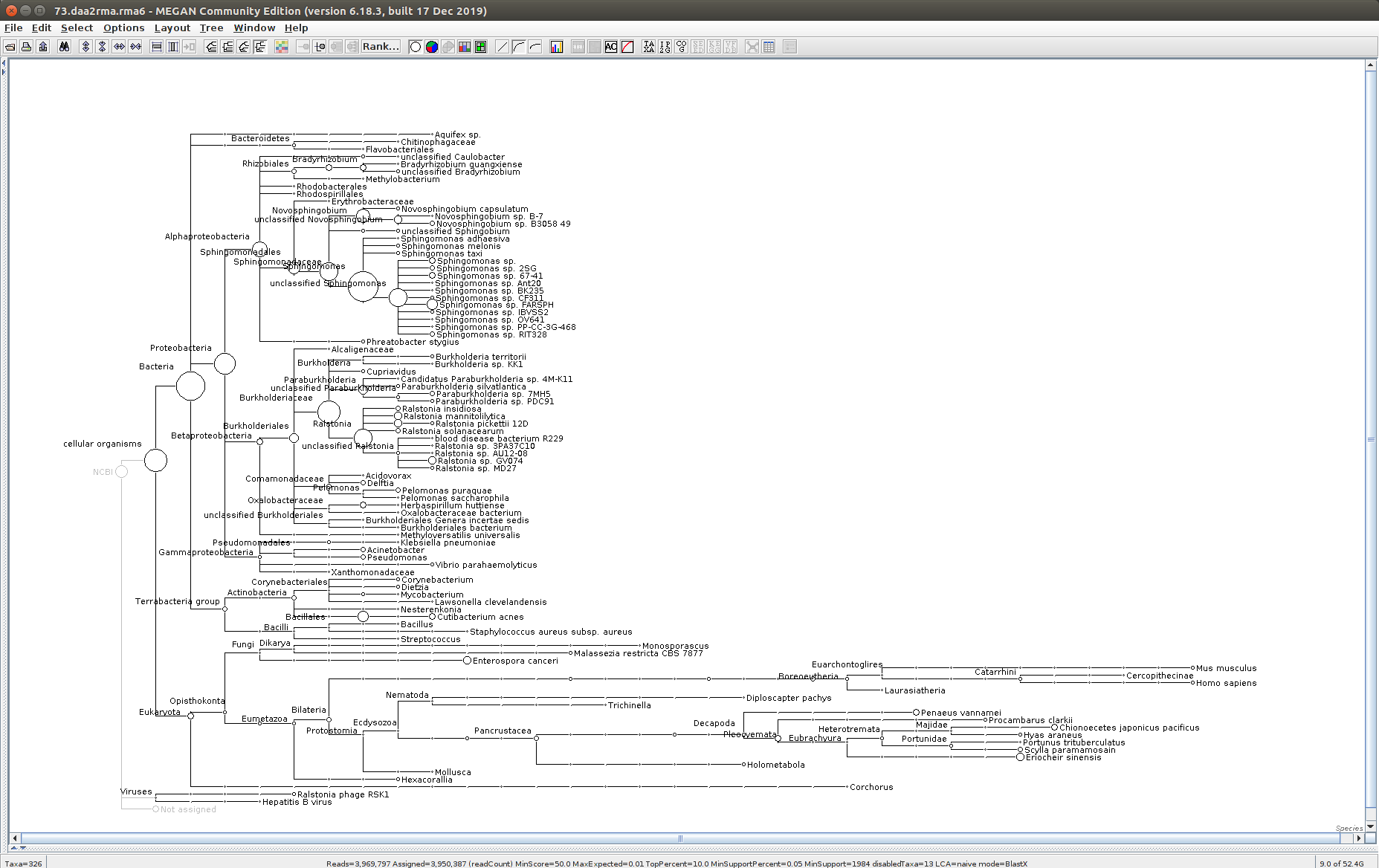
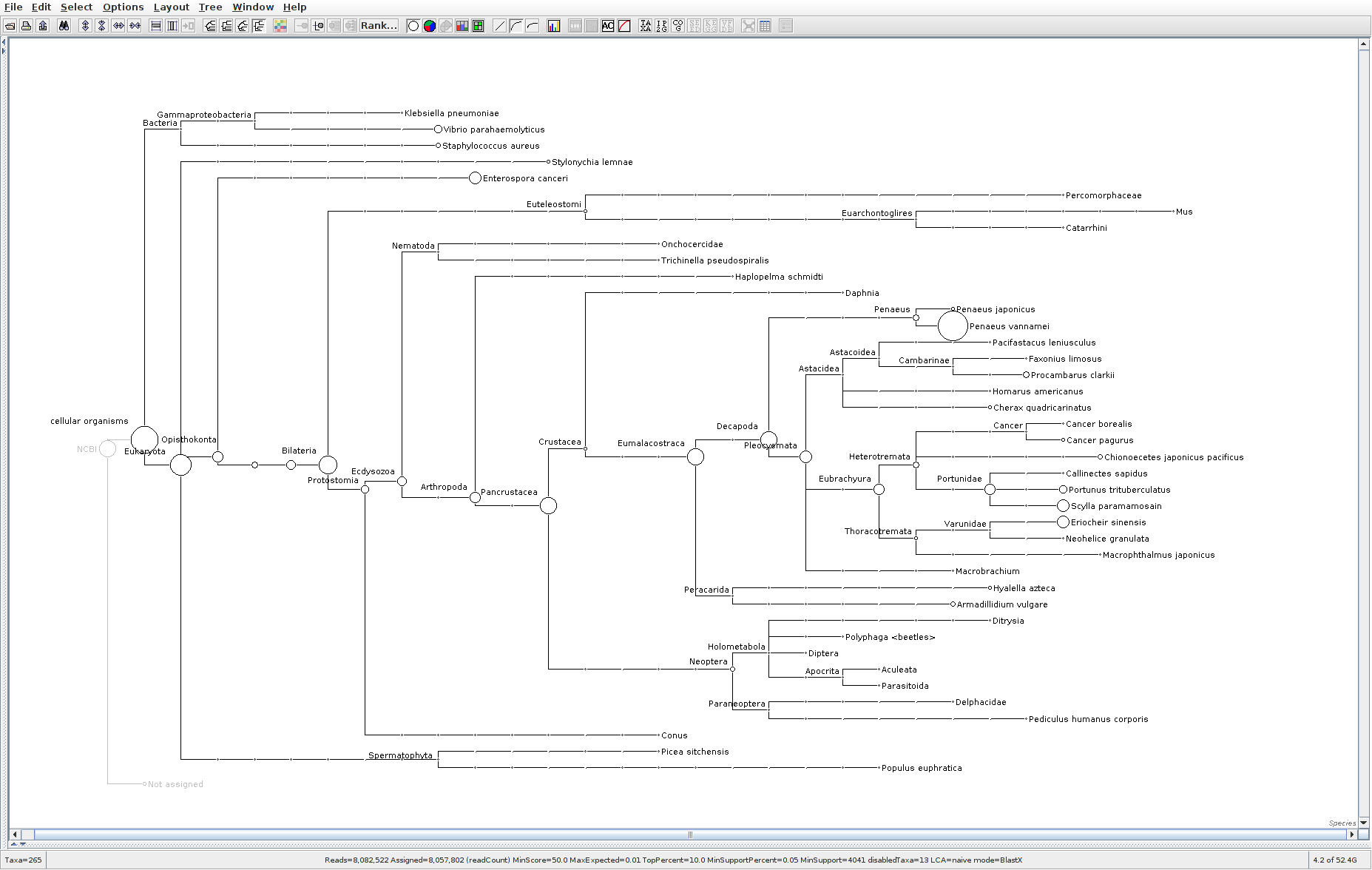
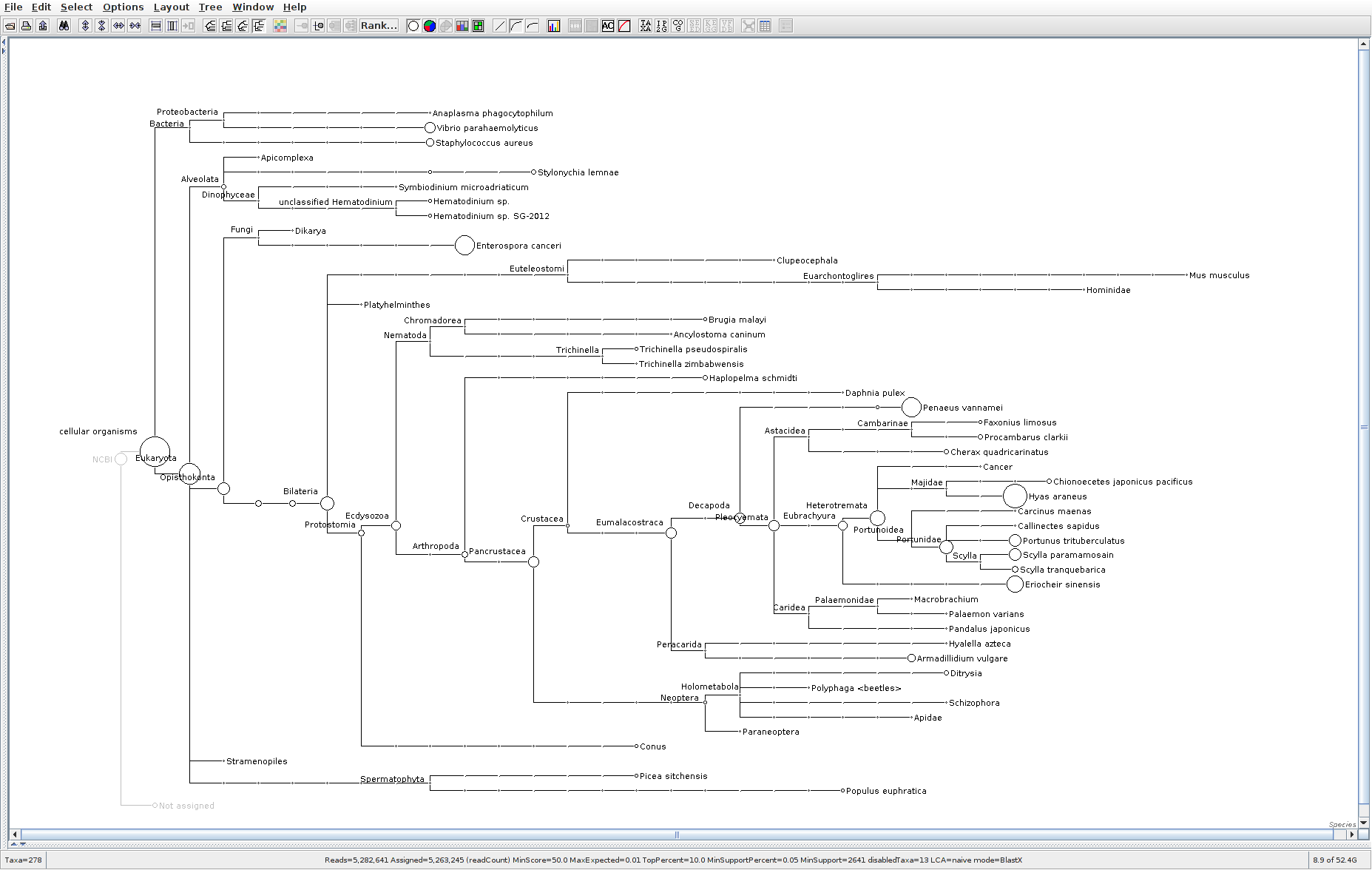
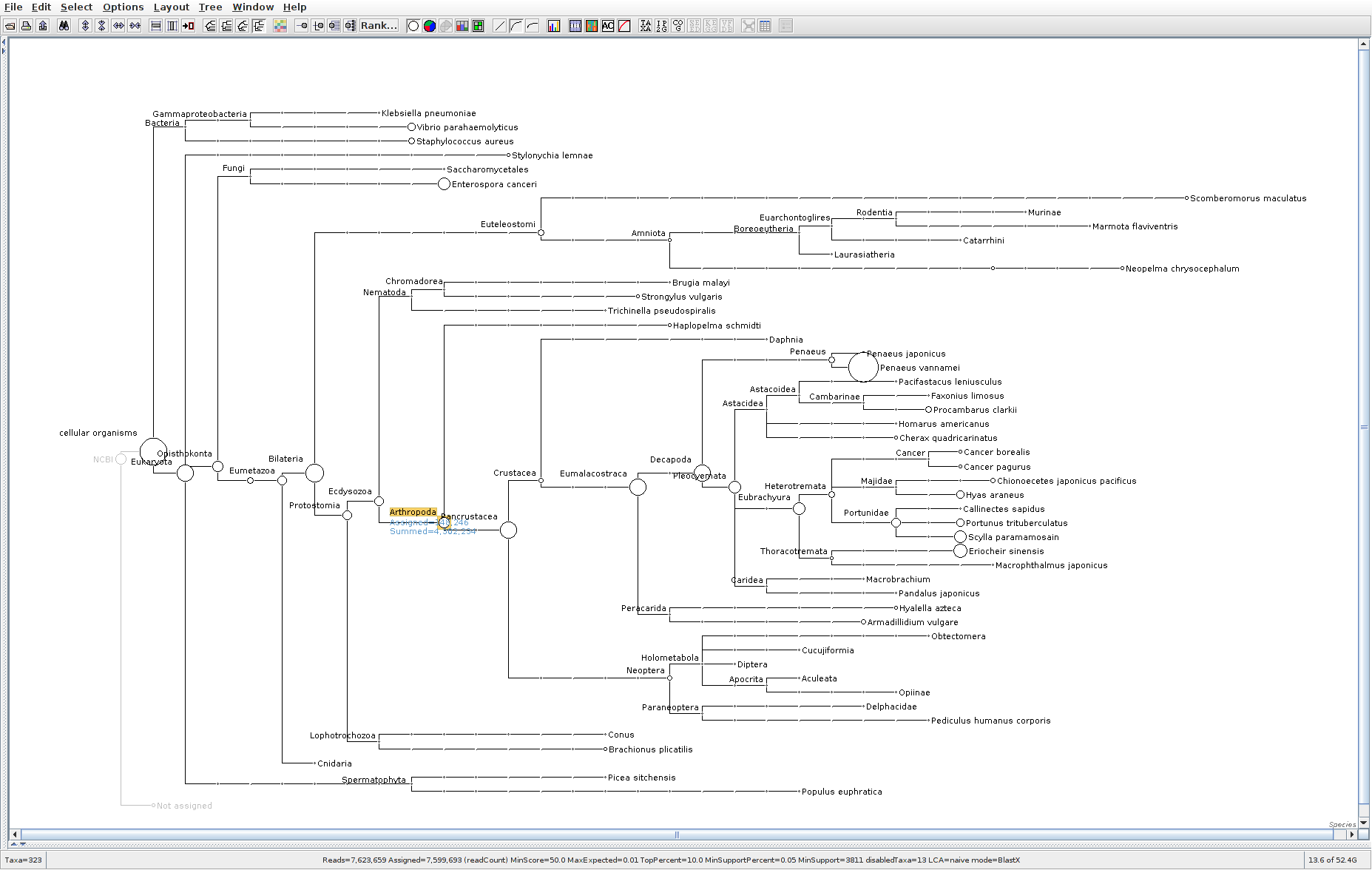
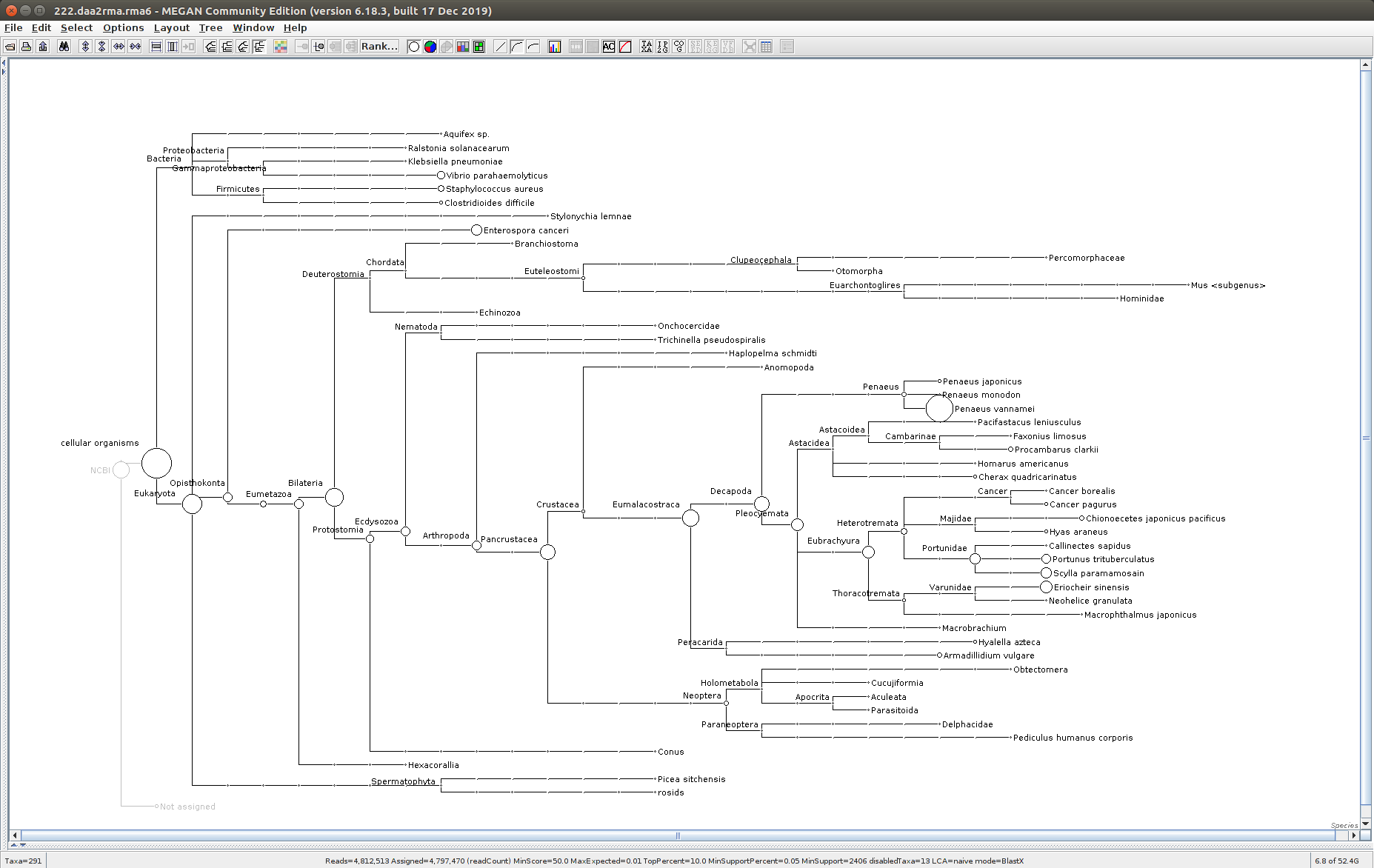
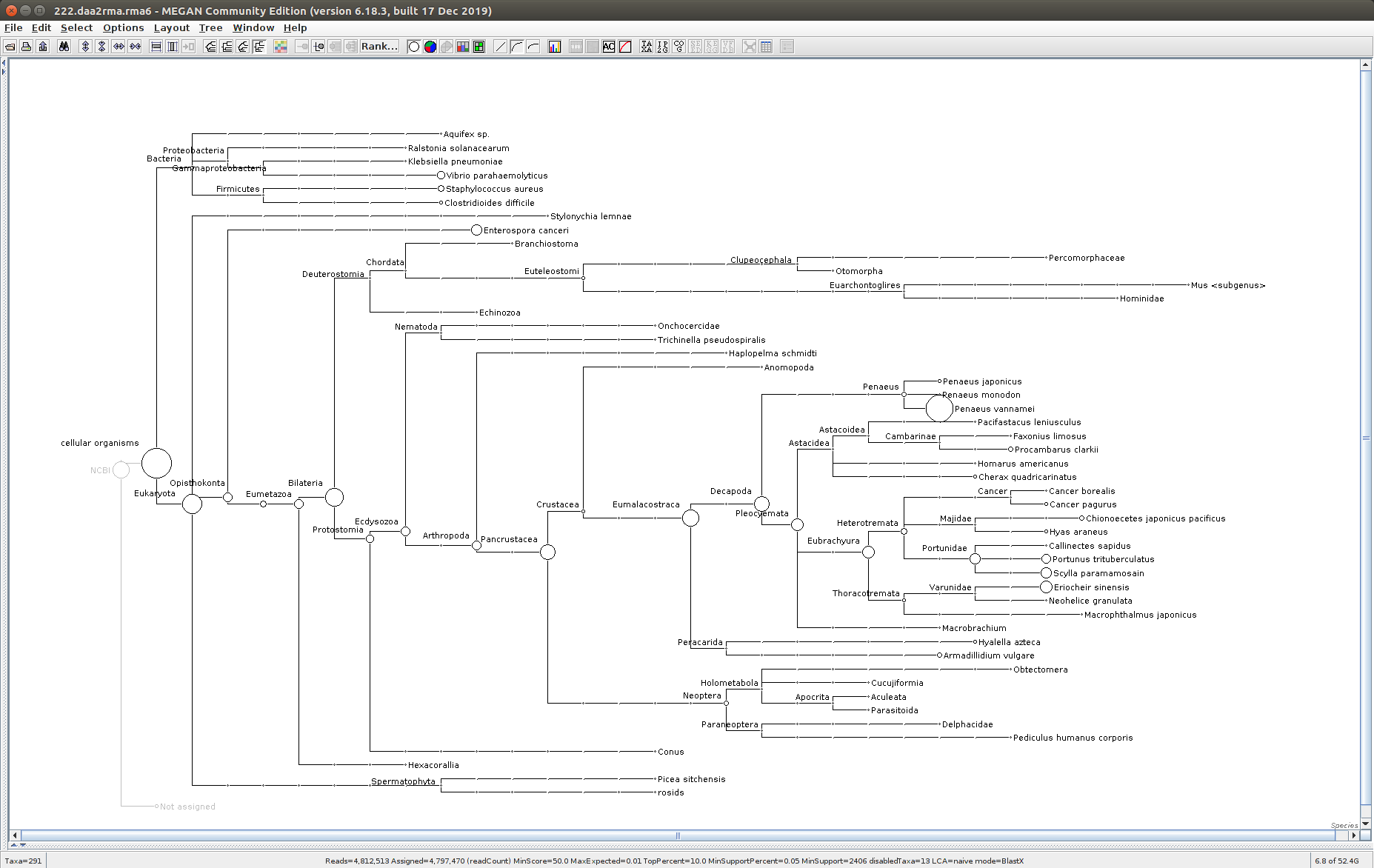
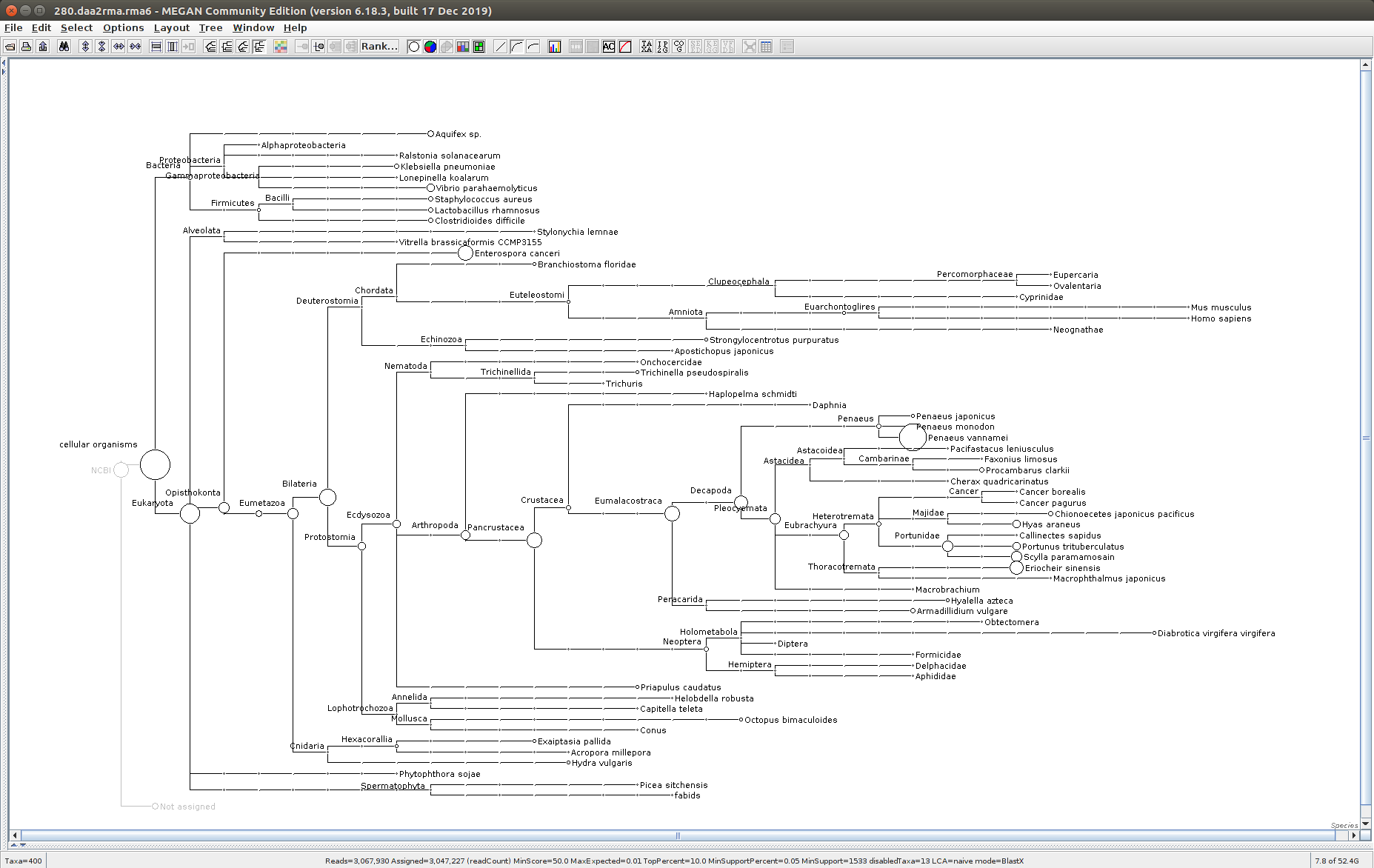
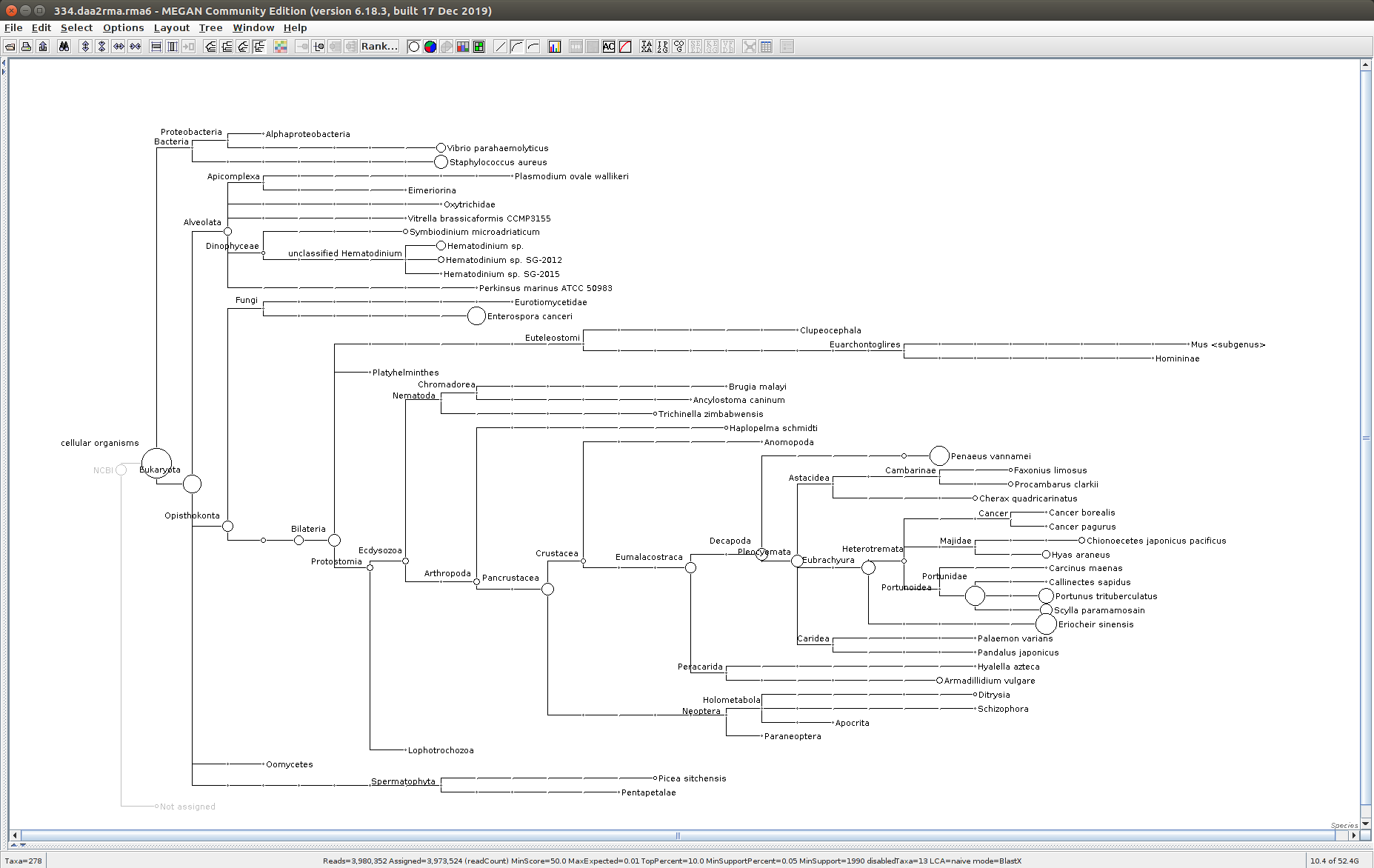
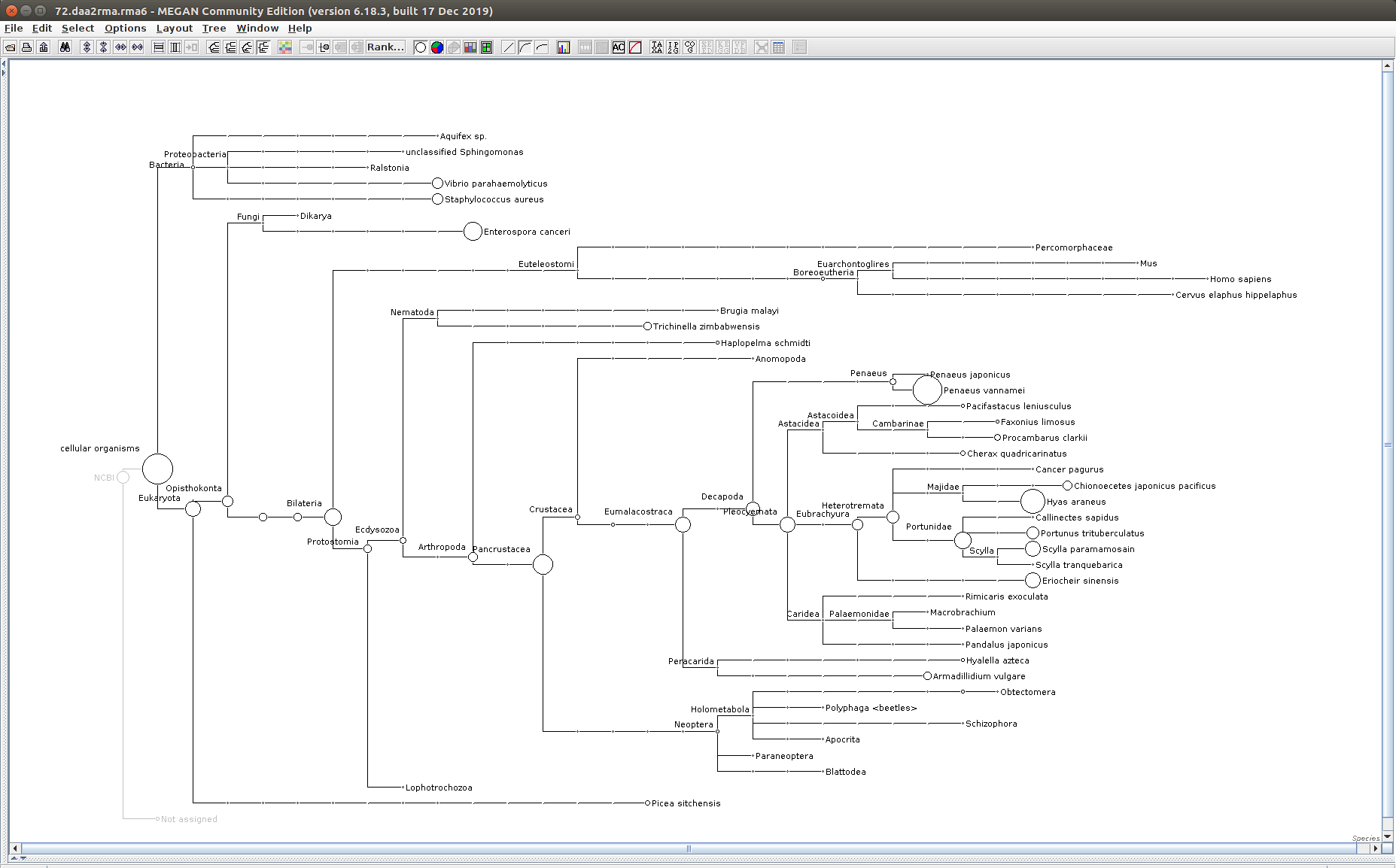
The taxonomic tree from each MEGAN6 RMA6 file is shown below. There are a couple of interesting things to notice from these:
Some samples have a high abundance of reads assigned to Bacteria. My guess is that this was due to a slight misstep in sampling, leading to collecting mostly sea water instead of mostly hemolymph. I say this because in these samples, there are still a large amount of Arthropoda reads, so it’s clear that some hemolymph was collected.
Most samples which should have reads assigned to Hematodinium (18 samples were considered “infected” via qPCR) do not have any reads assigned. In fact, only four samples ended up having Hematodinium reads extracted:
132
178
349
485
At this point in time, it’s not that big of a deal, since we’re currently just using this data to create an updated transcriptome for each of the two phyla.
- Many (most?) of the samples had a relatively high abundance of reads assigned to a microsporidian, Enterospora canceri, a known crab parasite. This is intriguing and not entirely sure what the implications are for analyzing the crab gene expression are. Also, it might be interesting to try to extract these reads and assemble a Enterospora canceri transcriptome…
Next up, creating some updated transcriptome assemblies/annotations for these two phyla.
Taxonomic Trees
113
118
127
132

151
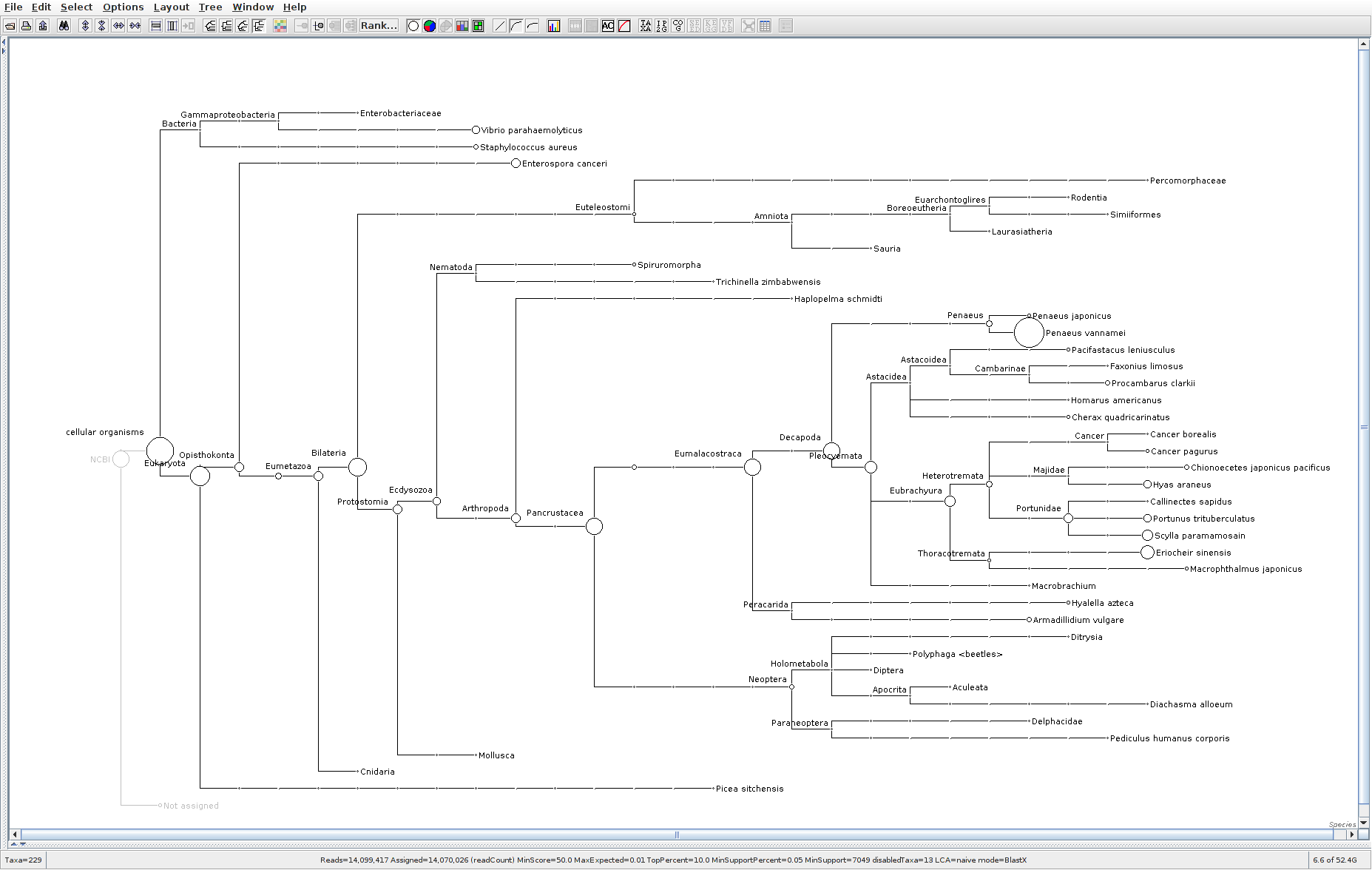
173
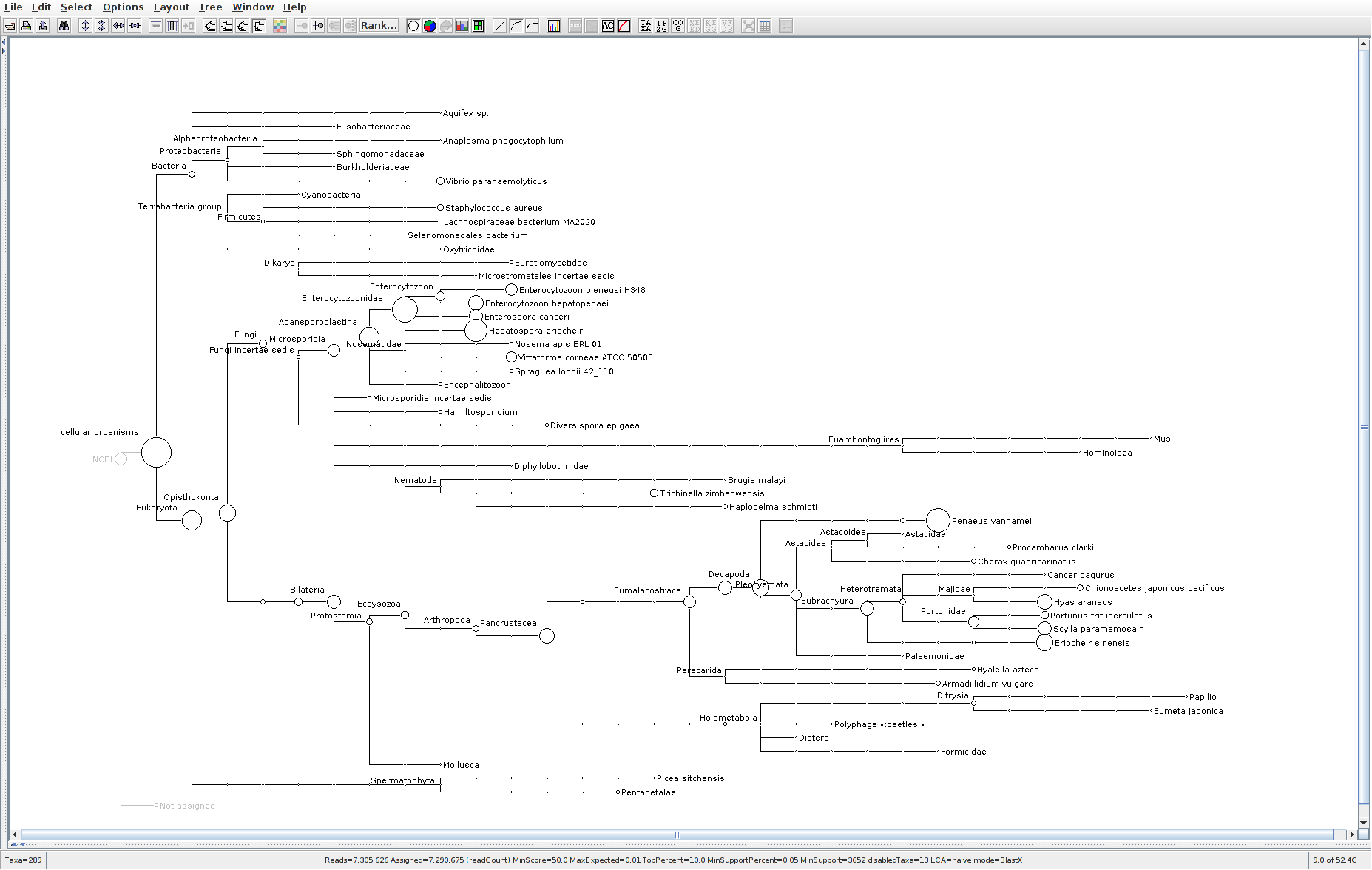
178
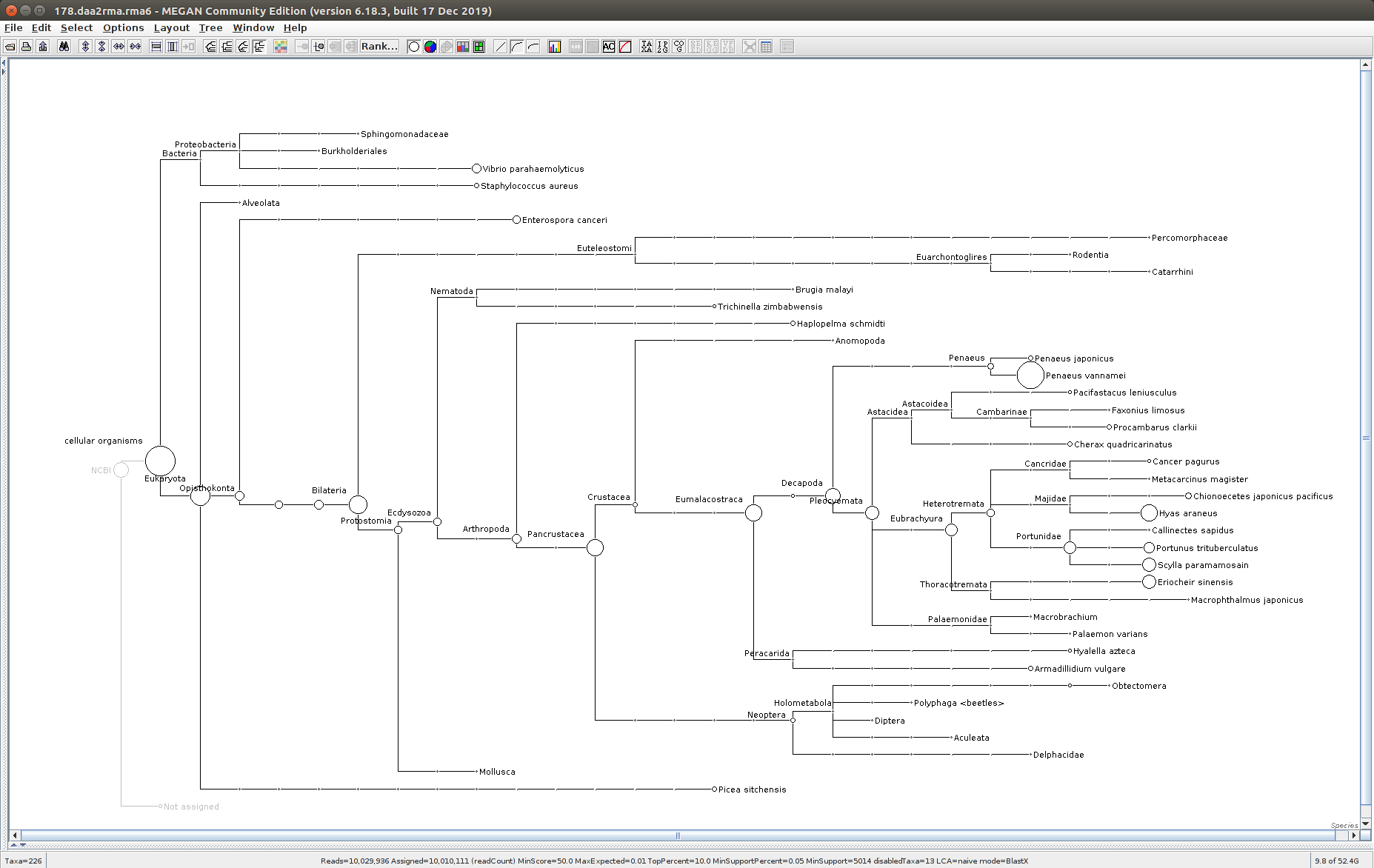
221
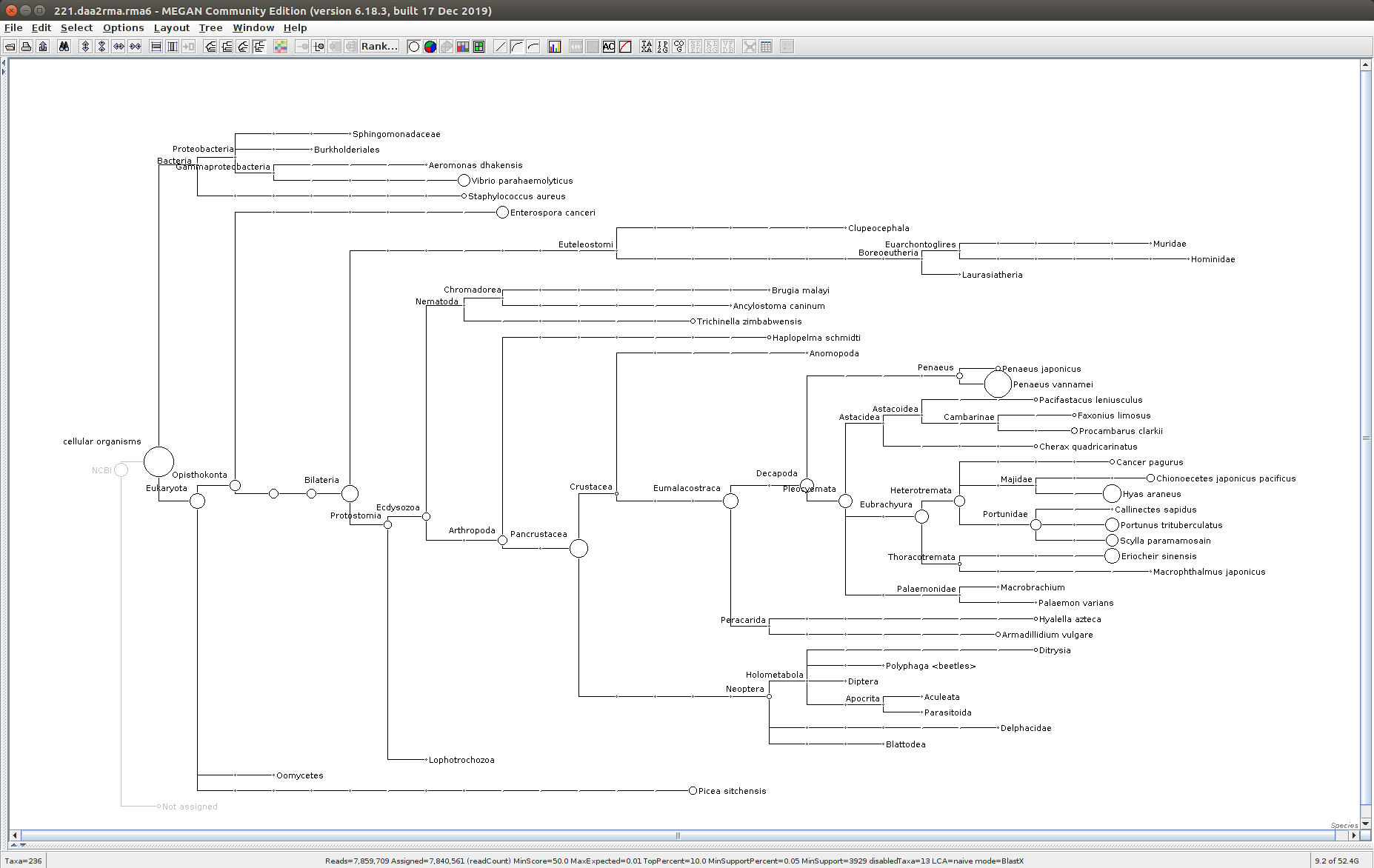
222
254
272

280
294

334
349
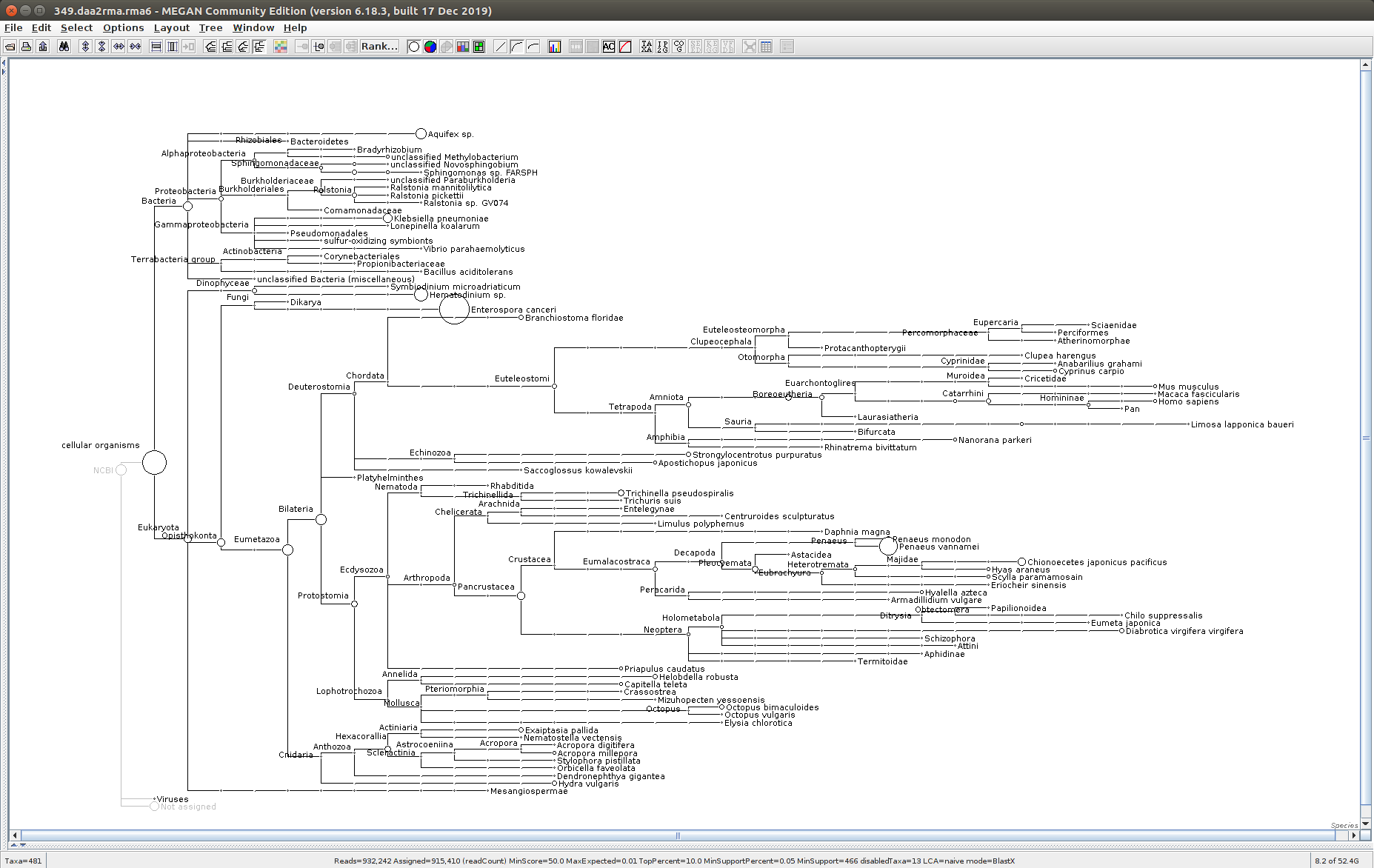
359
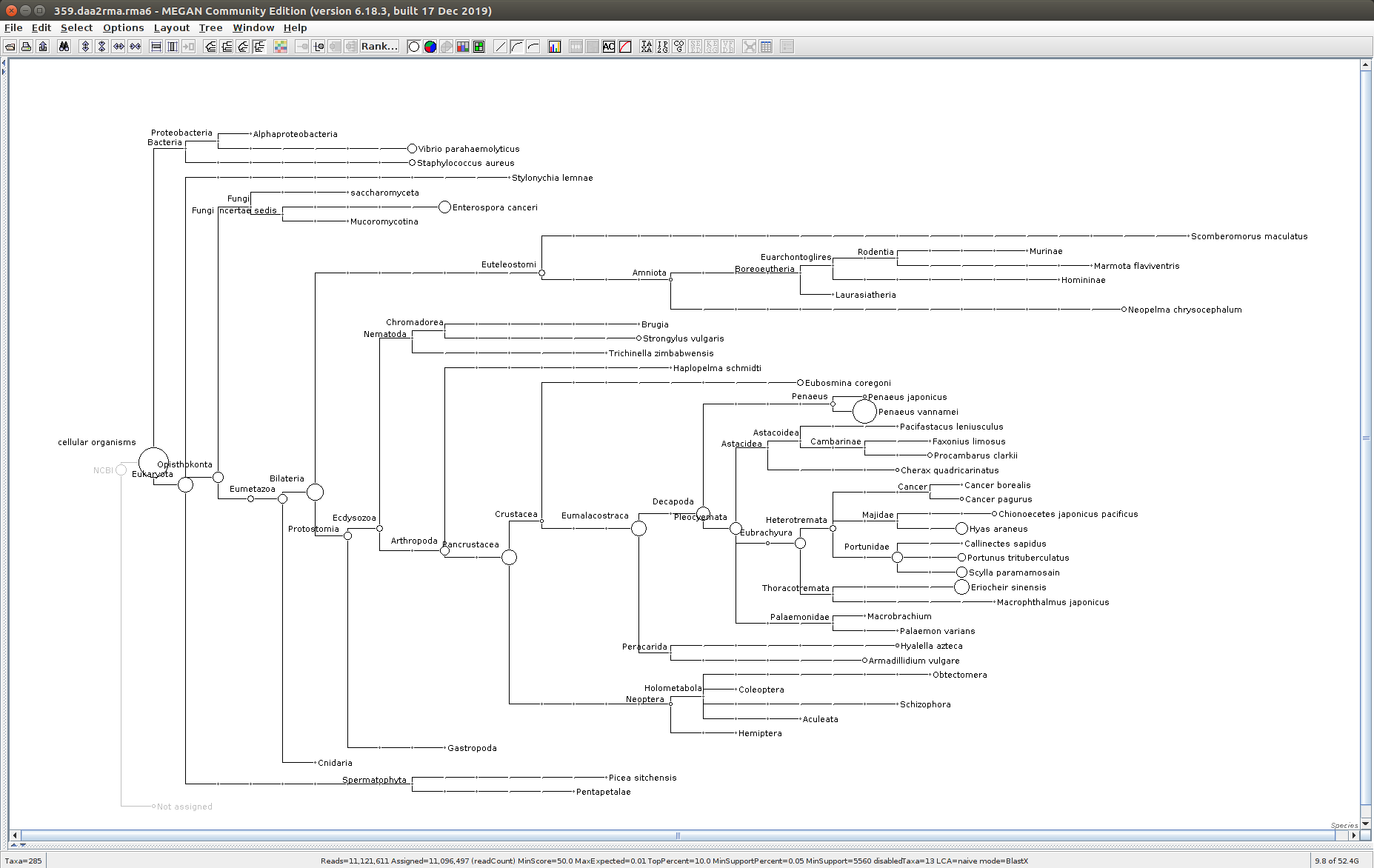
425
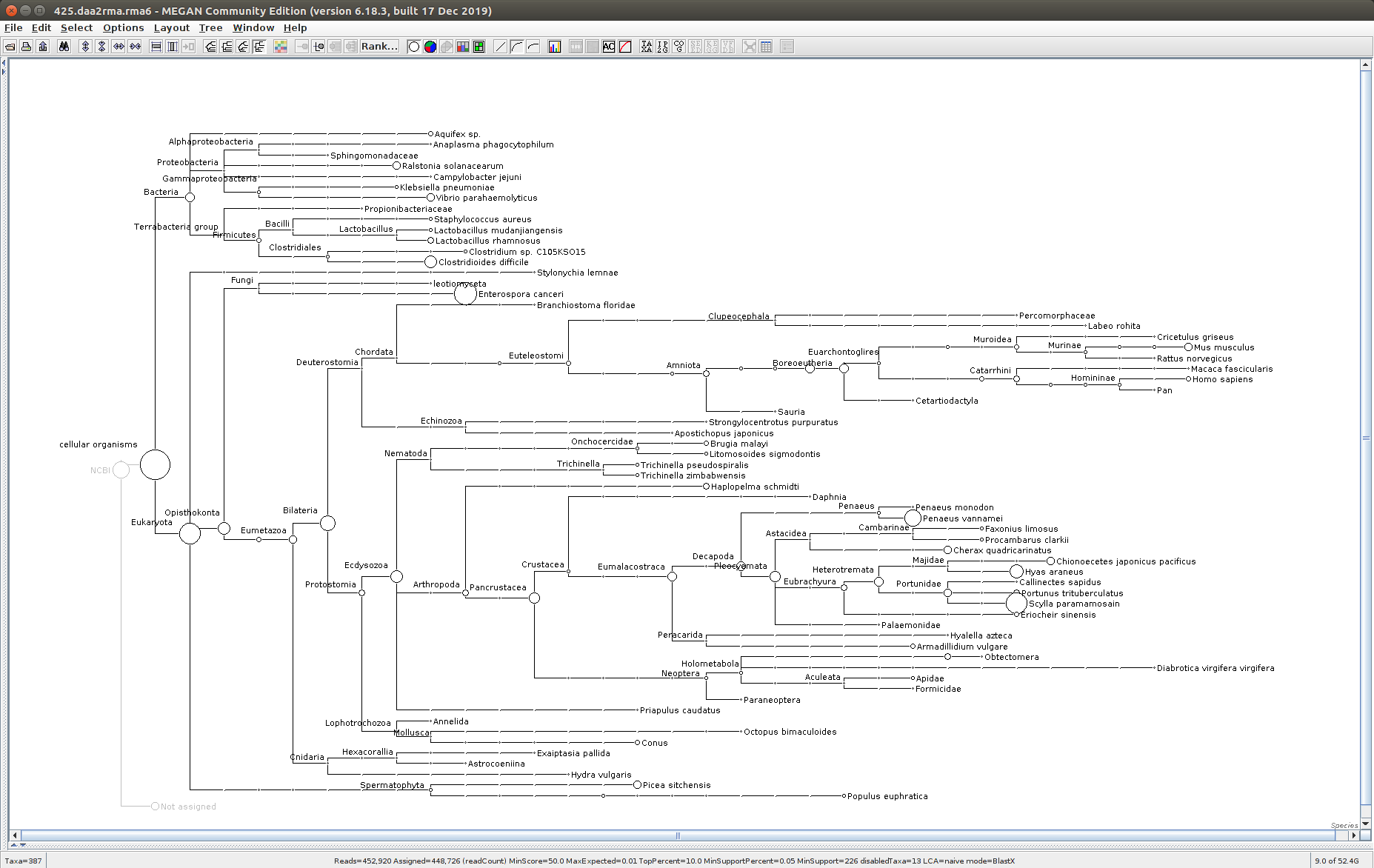
427
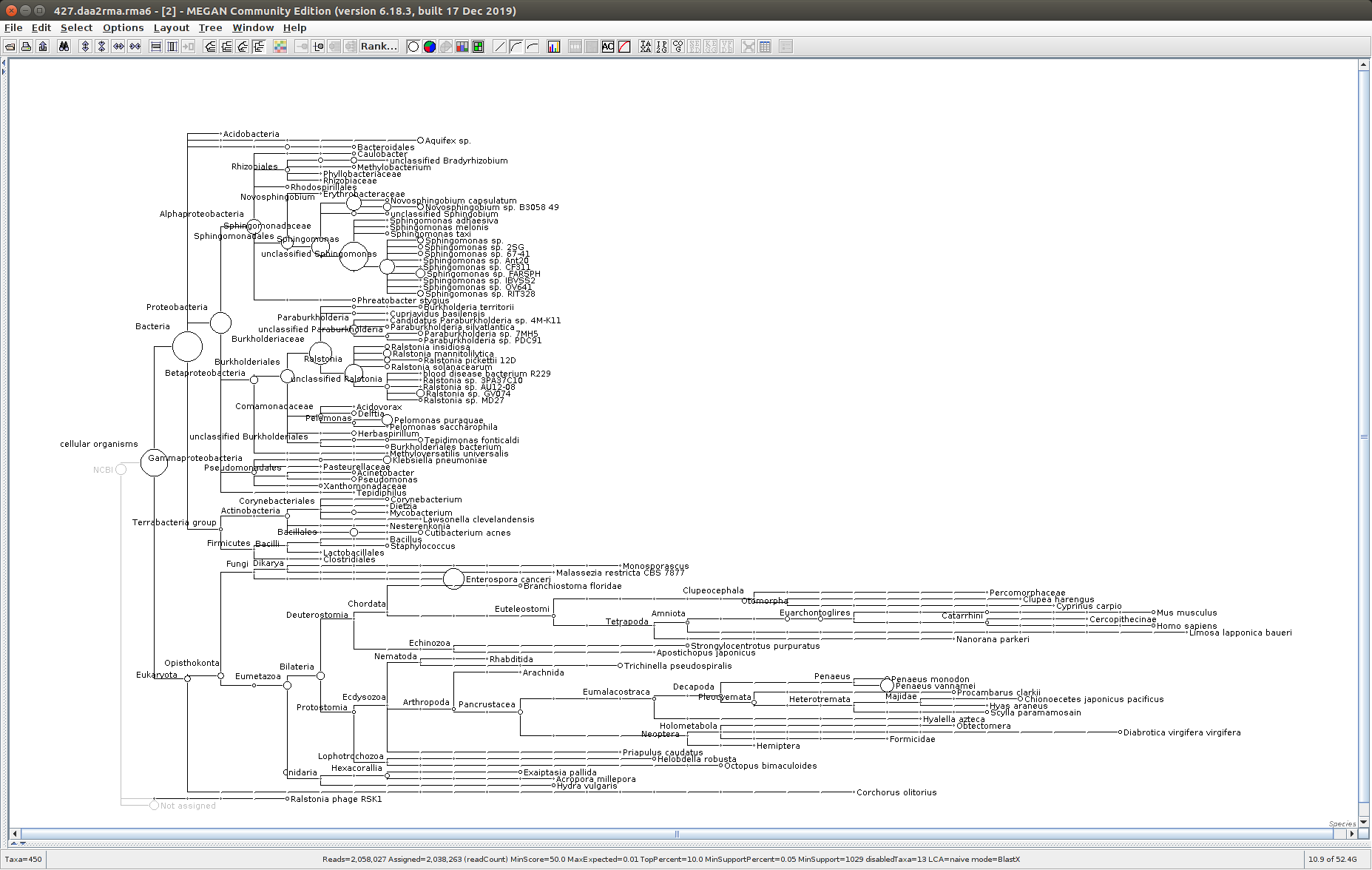
445
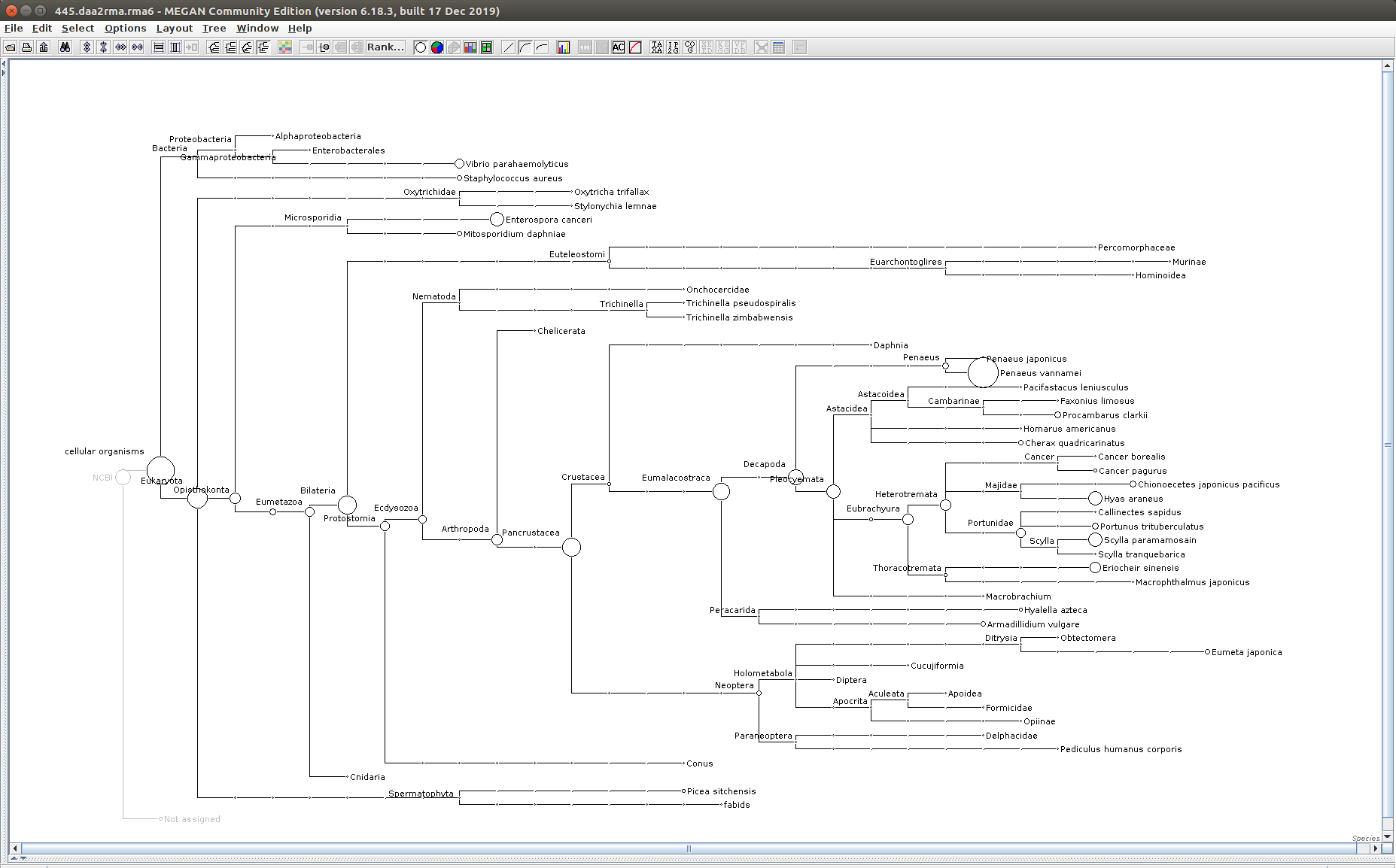
463
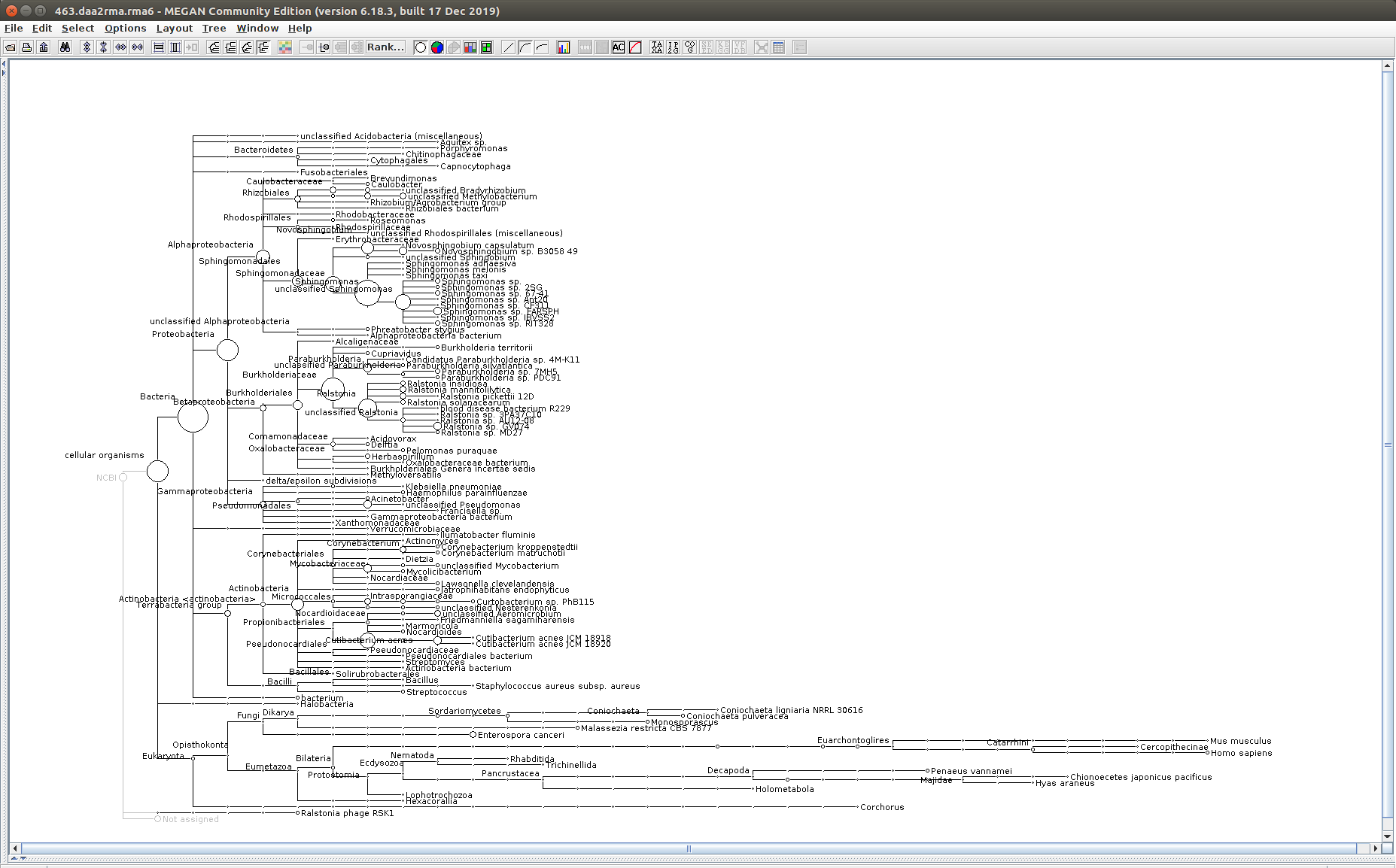
481
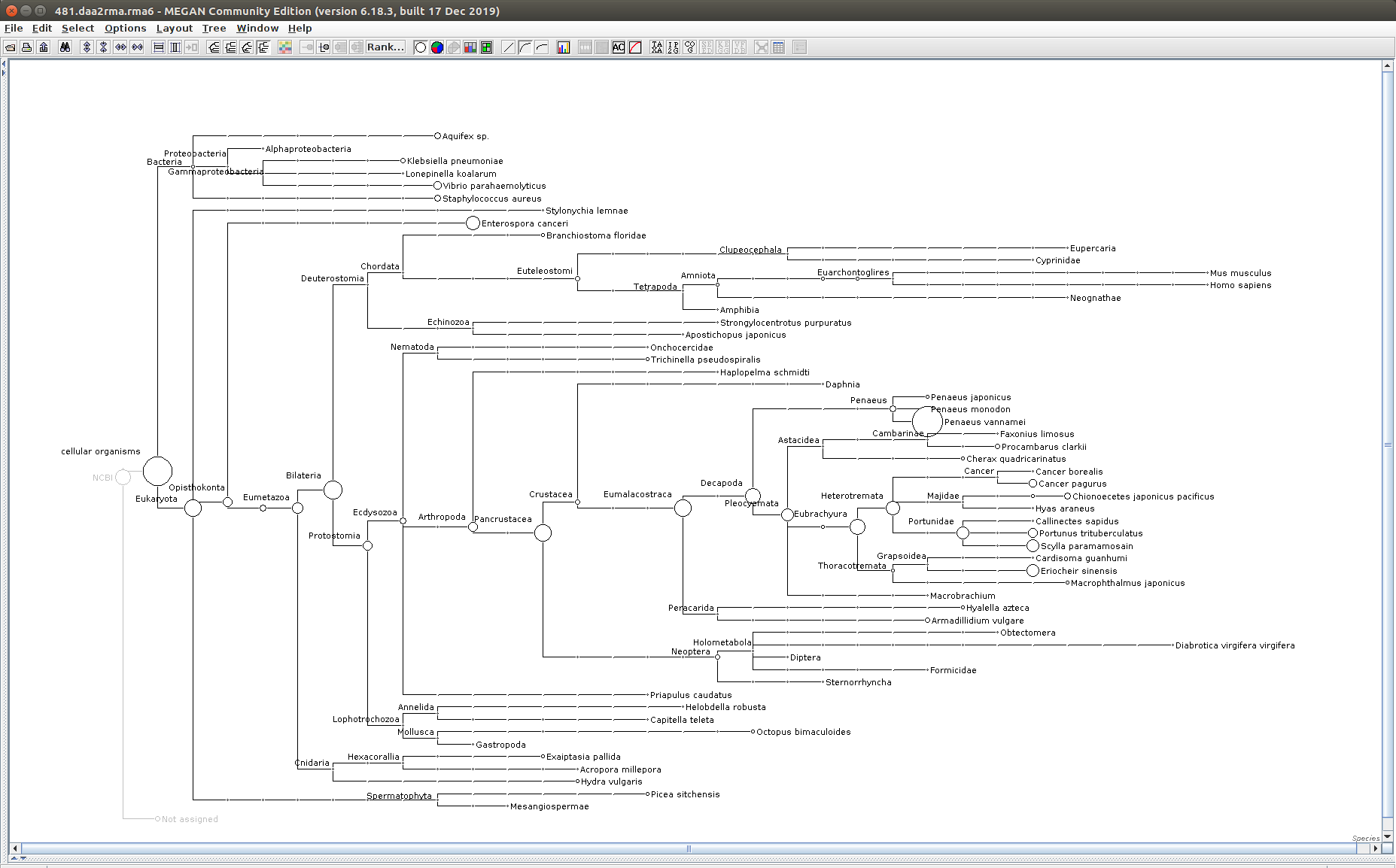
485
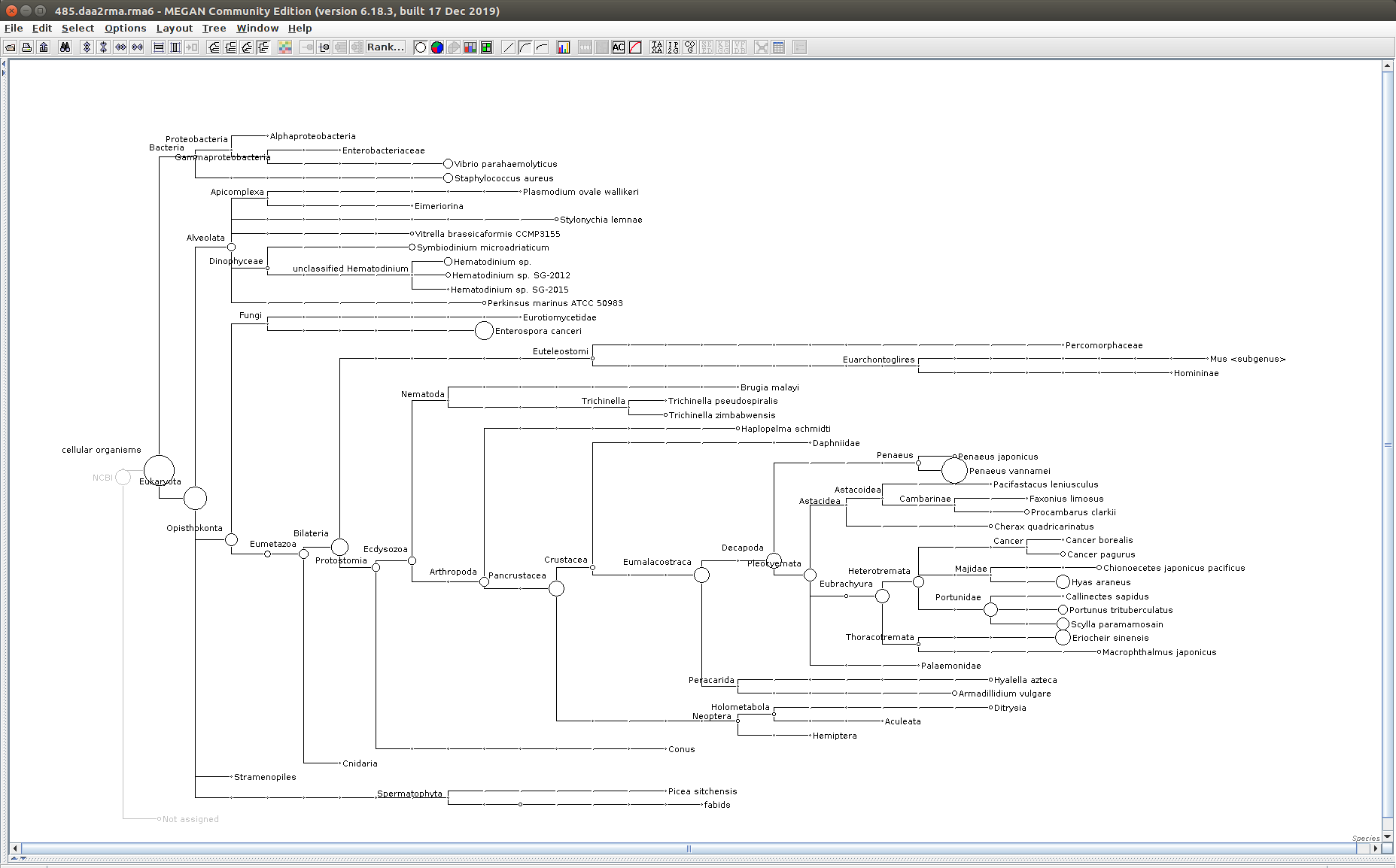
72
73