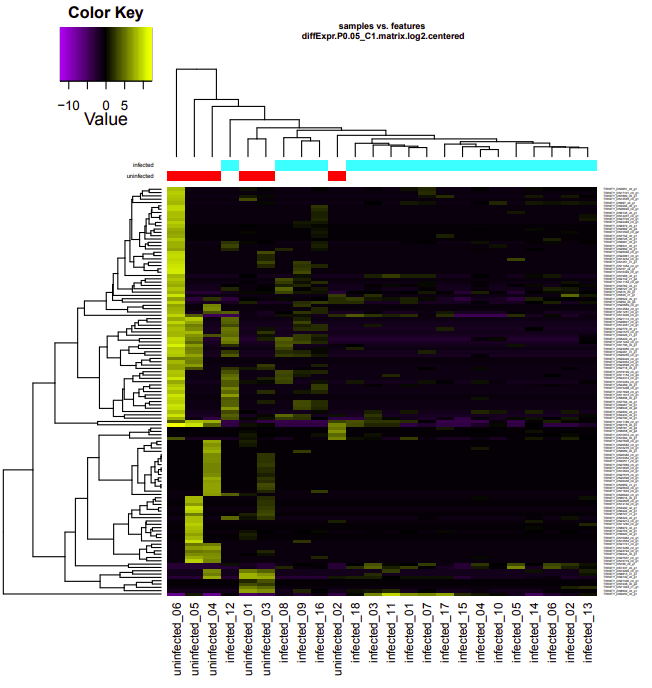
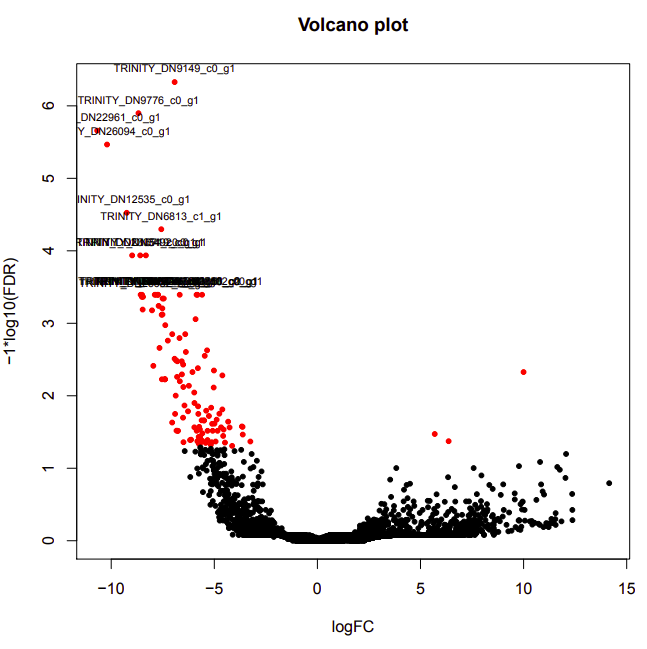
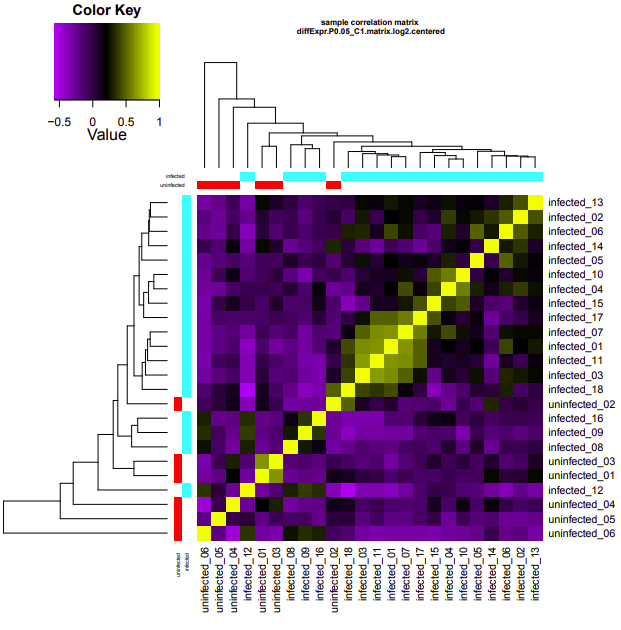
Per a Slack request, Steven asked me to take the Genewize RNAseq data (received 2020318) through edgeR. Ran the analysis using the Trinity differential expression pipeline:
Here’re the core input files used for this analysis:
Transcriptome: cbai_transcriptome_v1.5.fasta
MEGAN6 Arthropoda taxonomic reads: 20200413_C_bairdi_megan_reads/
The analyses will perform the following pairwise comparisons:
infected-uninfected
D9-D12
D9-D26
D12-D26
ambient-cold
ambient-warm
cold-warm
It will identify differentially expressed genes with >=2-fold log change in expression and a false discovery rate of <=0.05. Additionally, it will perform gene ontology (GO) enrichment analysis using GOseq.
As a brief aside, I’m pretty stoked about the SBATCH script below! It automates FastQ file selection for each comparison, creates appropriately named subdirectories and creates proper Trinity samples list file needed.
After running the DEG analysis, I “flattened” the enriched GO terms files for later use in R to map these GO terms to GOslims. That was run separately and the script is after the SBATCH script.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=cbai_DEG_basic
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=10-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20200422_cbai_DEG_basic_comparisons
# This is a script to identify differentially expressed genes (DEGs) in C.bairdi
# using pairwise comparisions of from just the "2020-GW" (i.e. just Genewiz) RNAseq data
# which has been taxonomically selected for all Arthropoda reads. See Sam's notebook from 20200419
# https://robertslab.github.io/sams-notebook
# Script will run Trinity's builtin differential gene expression analysis using:
# - Salmon alignment-free transcript abundance estimation
# - edgeR
# Cutoffs of 2-fold difference in expression and FDR of <=0.05.
###################################################################################
# These variables need to be set by user
fastq_dir="/gscratch/srlab/sam/data/C_bairdi/RNAseq/"
fasta_prefix="20200408.C_bairdi.megan.Trinity"
transcriptome_dir="/gscratch/srlab/sam/data/C_bairdi/transcriptomes"
trinotate_feature_map="${transcriptome_dir}/20200409.cbai.trinotate.annotation_feature_map.txt"
go_annotations="${transcriptome_dir}/20200409.cbai.trinotate.go_annotations.txt"
# Array of the various comparisons to evaluate
# Each condition in each comparison should be separated by a "-"
comparisons_array=(
infected-uninfected \
D9-D12 \
D9-D26 \
D12-D26 \
ambient-cold \
ambient-warm \
cold-warm
)
# Functions
# Expects input (i.e. "$1") to be in the following format:
# e.g. 20200413.C_bairdi.113.D9.uninfected.cold.megan_R2.fq
get_day () { day=$(echo "$1" | awk -F"." '{print $4}'); }
get_inf () { inf=$(echo "$1" | awk -F"." '{print $5}'); }
get_temp () { temp=$(echo "$1" | awk -F"." '{print $6}'); }
###################################################################################
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
wd="$(pwd)"
threads=28
## Designate input file locations
transcriptome="${transcriptome_dir}/${fasta_prefix}.fasta"
fasta_seq_lengths="${transcriptome_dir}/${fasta_prefix}.fasta.seq_lens"
gene_map="${transcriptome_dir}/${fasta_prefix}.fasta.gene_trans_map"
transcriptome="${transcriptome_dir}/${fasta_prefix}.fasta"
# Standard output/error files
diff_expr_stdout="diff_expr_stdout.txt"
diff_expr_stderr="diff_expr_stderr.txt"
matrix_stdout="matrix_stdout.txt"
matrix_stderr="matrix_stderr.txt"
salmon_stdout="salmon_stdout.txt"
salmon_stderr="salmon_stderr.txt"
tpm_length_stdout="tpm_length_stdout.txt"
tpm_length_stderr="tpm_length_stderr.txt"
trinity_DE_stdout="trinity_DE_stdout.txt"
trinity_DE_stderr="trinity_DE_stderr.txt"
edgeR_dir=""
#programs
trinity_home=/gscratch/srlab/programs/trinityrnaseq-v2.9.0
trinity_annotate_matrix="${trinity_home}/Analysis/DifferentialExpression/rename_matrix_feature_identifiers.pl"
trinity_abundance=${trinity_home}/util/align_and_estimate_abundance.pl
trinity_matrix=${trinity_home}/util/abundance_estimates_to_matrix.pl
trinity_DE=${trinity_home}/Analysis/DifferentialExpression/run_DE_analysis.pl
diff_expr=${trinity_home}/Analysis/DifferentialExpression/analyze_diff_expr.pl
trinity_tpm_length=${trinity_home}/util/misc/TPM_weighted_gene_length.py
# Loop through each comparison
# Will create comparison-specific direcctories and copy
# appropriate FastQ files for each comparison.
# After file transfer, will create necessary sample list file for use
# by Trinity for running differential gene expression analysis and GO enrichment.
for comparison in "${!comparisons_array[@]}"
do
# Assign variables
cond1_count=0
cond2_count=0
comparison=${comparisons_array[${comparison}]}
comparison_dir=${wd}/${comparison}
salmon_gene_matrix=${comparison_dir}/salmon.gene.TMM.EXPR.matrix
salmon_iso_matrix=${comparison_dir}/salmon.isoform.TMM.EXPR.matrix
samples=${comparison_dir}${comparison}.samples.txt
# Extract each comparison from comparisons array
# Conditions must be separated by a "-"
cond1=$(echo "${comparison}" | awk -F"-" '{print $1}')
cond2=$(echo "${comparison}" | awk -F"-" '{print $2}')
mkdir --parents "${comparison}"
cd "${comparison}" || exit
# Series of if statements to identify which FastQ files to rsync to working directory
if [[ "${comparison}" == "infected-uninfected" ]]; then
rsync --archive --verbose ${fastq_dir}*.fq .
fi
if [[ "${comparison}" == "D9-D12" ]]; then
for fastq in "${fastq_dir}"*.fq
do
get_day "${fastq}"
if [[ "${day}" == "D9" || "${day}" == "D12" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
if [[ "${comparison}" == "D9-D26" ]]; then
for fastq in "${fastq_dir}"*.fq
do
get_day "${fastq}"
if [[ "${day}" == "D9" || "${day}" == "D26" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
if [[ "${comparison}" == "D12-D26" ]]; then
for fastq in "${fastq_dir}"*.fq
do
get_day "${fastq}"
if [[ "${day}" == "D12" || "${day}" == "D26" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
if [[ "${comparison}" == "ambient-cold" ]]; then
#statements
for fastq in "${fastq_dir}"*.fq
do
get_temp "${fastq}"
if [[ "${temp}" == "ambient" || "${temp}" == "cold" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
if [[ "${comparison}" == "ambient-warm" ]]; then
for fastq in "${fastq_dir}"*.fq
do
get_temp "${fastq}"
if [[ "${temp}" == "ambient" || "${temp}" == "warm" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
if [[ "${comparison}" == "cold-warm" ]]; then
for fastq in "${fastq_dir}"*.fq
do
get_temp "${fastq}"
if [[ "${temp}" == "cold" || "${temp}" == "warm" ]]; then
rsync --archive --verbose "${fastq}" .
fi
done
fi
# Create reads array
# Paired reads files will be sequentially listed in array (e.g. 111_R1 111_R2)
reads_array=(*.fq)
echo ""
echo "Created reads_array"
# Loop to create sample list file
# Sample file list is tab-delimited like this:
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
# Increment by 2 to process next pair of FastQ files
for (( i=0; i<${#reads_array[@]} ; i+=2 ))
do
echo ""
echo "Evaluating ${reads_array[i]} and ${reads_array[i+1]}"
get_day "${reads_array[i]}"
get_inf "${reads_array[i]}"
get_temp "${reads_array[i]}"
echo ""
echo "Got day (${day}), infection status (${inf}), and temp (${temp})."
echo ""
echo "Condition 1 is: ${cond1}"
echo "condition 2 is: ${cond2}"
# Evaluate specified treatment conditions and format sample file list appropriately.
if [[ "${cond1}" == "${day}" || "${cond1}" == "${inf}" || "${cond1}" == "${temp}" ]]; then
cond1_count=$((cond1_count+1))
echo ""
echo "Condition 1 evaluated."
# Create tab-delimited samples file.
printf "%s\t%s%02d\t%s\t%s\n" "${cond1}" "${cond1}_" "${cond1_count}" "${comparison_dir}${reads_array[i]}" "${comparison_dir}${reads_array[i+1]}" \
>> "${samples}"
elif [[ "${cond2}" == "${day}" || "${cond2}" == "${inf}" || "${cond2}" == "${temp}" ]]; then
cond2_count=$((cond2_count+1))
echo ""
echo "Condition 2 evaluated."
# Create tab-delimited samples file.
printf "%s\t%s%02d\t%s\t%s\n" "${cond2}" "${cond2}_" "${cond2_count}" "${comparison_dir}${reads_array[i]}" "${comparison_dir}${reads_array[i+1]}" \
>> "${samples}"
fi
# Copy sample list file to transcriptome directory
cp "${samples}" "${transcriptome_dir}"
done
echo "Created ${comparison} sample list file."
# Create directory/sample list for ${trinity_matrix} command
trin_matrix_list=$(awk '{printf "%s%s", $2, "/quant.sf " }' "${samples}")
# Determine transcript abundances using Salmon alignment-free
# abundance estimate.
${trinity_abundance} \
--output_dir "${comparison_dir}" \
--transcripts ${transcriptome} \
--seqType fq \
--samples_file "${samples}" \
--est_method salmon \
--aln_method bowtie2 \
--gene_trans_map "${gene_map}" \
--prep_reference \
--thread_count "${threads}" \
1> "${comparison_dir}"${salmon_stdout} \
2> "${comparison_dir}"${salmon_stderr}
# Convert abundance estimates to matrix
${trinity_matrix} \
--est_method salmon \
--gene_trans_map ${gene_map} \
--out_prefix salmon \
--name_sample_by_basedir \
${trin_matrix_list} \
1> ${matrix_stdout} \
2> ${matrix_stderr}
# Integrate Trinotate functional annotations
"${trinity_annotate_matrix}" \
"${trinotate_feature_map}" \
salmon.gene.counts.matrix \
> salmon.gene.counts.annotated.matrix
# Generate weighted gene lengths
"${trinity_tpm_length}" \
--gene_trans_map "${gene_map}" \
--trans_lengths "${fasta_seq_lengths}" \
--TPM_matrix "${salmon_iso_matrix}" \
> Trinity.gene_lengths.txt \
2> ${tpm_length_stderr}
# Differential expression analysis
# Utilizes edgeR.
# Needs to be run in same directory as transcriptome.
cd ${transcriptome_dir} || exit
${trinity_DE} \
--matrix "${comparison_dir}salmon.gene.counts.matrix" \
--method edgeR \
--samples_file "${samples}" \
1> ${trinity_DE_stdout} \
2> ${trinity_DE_stderr}
mv edgeR* "${comparison_dir}"
# Run differential expression on edgeR output matrix
# Set fold difference to 2-fold (ie. -C 1 = 2^1)
# P value <= 0.05
# Has to run from edgeR output directory
# Pulls edgeR directory name and removes leading ./ in find output
# Using find is required because edgeR names directory using PID
# and I don't know how to find that out
cd "${comparison_dir}" || exit
edgeR_dir=$(find . -type d -name "edgeR*" | sed 's%./%%')
cd "${edgeR_dir}" || exit
mv "${transcriptome_dir}/${trinity_DE_stdout}" .
mv "${transcriptome_dir}/${trinity_DE_stderr}" .
${diff_expr} \
--matrix "${salmon_gene_matrix}" \
--samples "${samples}" \
--examine_GO_enrichment \
--GO_annots "${go_annotations}" \
--include_GOplot \
--gene_lengths "${comparison_dir}Trinity.gene_lengths.txt" \
-C 1 \
-P 0.05 \
1> ${diff_expr_stdout} \
2> ${diff_expr_stderr}
cd "${wd}" || exit
doneFlatten enriched GO terms file (GitHub):
#!/bin/bash
#############################################################
# Script to "flatten" Trinity edgeR GOseq enrichment format
# so each line contains a single gene/transcript ID
# and associated GO term
#############################################################
# Enable globstar for recursive searching
shopt -s globstar
# Declare variables
output_file=""
wd=$(pwd)
# Input file
## Expects Trinity edgeR GOseq enrichment format:
## category over_represented_pvalue under_represented_pvalue numDEInCat numInCat term ontology over_represented_FDR go_term gene_ids
## Field 10 (gene_ids) contains comma separated gene_ids that fall in the given GO term in the "category" column
for goseq in **/*UP.subset*.enriched
do
# Capture path to file
dir=${goseq%/*}
cd "${dir}" || exit
tmp_file=$(mktemp)
# Count lines in file
linecount=$(cat "${goseq}" | wc -l)
# If file is not empty
if (( "${linecount}" > 1 ))
then
output_file="${goseq}.flattened"
# 1st: Convert comma-delimited gene IDs in column 10 to tab-delimited
# Also, set output (OFS) to be tab-delimited
# 2nd: Convert spaces to underscores and keep output as tab-delimited
# 3rd: Sort on Trinity IDs (column 10) and keep only uniques
awk 'BEGIN{FS="\t";OFS="\t"} {gsub(/, /, "\t", $10); print}' "${goseq}" \
| awk 'BEGIN{F="\t";OFS="\t"} NR==1; NR > 1 {gsub(/ /, "_", $0); print}' \
> "${tmp_file}"
# Identify the first line number which contains a gene_id
begin_goterms=$(grep --line-number "TRINITY" "${tmp_file}" \
| awk '{for (i=1;i<=NF;i++) if($i ~/TRINITY/) print i}' \
| sort --general-numeric-sort --unique | head -n1)
# "Unfolds" gene_ids to a single gene_id per row
while read -r line
do
# Capture the length of the longest row
max_field=$(echo "$line" | awk -F "\t" '{print NF}')
# Retain the first 8 fields (i.e. categories)
fixed_fields=$(echo "$line" | cut -f1-8)
# Since not all the lines contain the same number of fields (e.g. may not have GO terms),
# evaluate the number of fields in each line to determine how to handle current line.
# If the value in max_field is less than the field number where the GO terms begin,
# then just print the current line (%s) followed by a newline (\n).
if (( "$max_field" < "$begin_goterms" ))
then
printf "%s\n" "$line"
else goterms=$(echo "$line" | cut -f"$begin_goterms"-"$max_field")
# Assign values in the variable "goterms" to a new indexed array (called "array"),
# with tab delimiter (IFS=$'\t')
IFS=$'\t' read -r -a array <<<"$goterms"
# Iterate through each element of the array.
# Print the first n fields (i.e. the fields stored in "fixed_fields") followed by a tab (%s\t).
# Print the current element in the array (i.e. the current GO term) followed by a new line (%s\n).
for element in "${!array[@]}"
do
printf "%s\t%s\n" "$fixed_fields" "${array[$element]}"
done
fi
done < "${tmp_file}" > "${output_file}"
fi
# Cleanup
rm "${tmp_file}"
cd "${wd}" || exit
doneRESULTS
Took about 17.5hrs to run:

Output folder:
D9-D12
Up-regulated genes:
salmon.gene.counts.matrix.D12_vs_D9.edgeR.DE_results.P0.05_C1.D9-UP.subset
salmon.gene.counts.matrix.D12_vs_D9.edgeR.DE_results.P0.05_C1.D12-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.D12_vs_D9.edgeR.DE_results.P0.05_C1.D12-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.D12_vs_D9.edgeR.DE_results.P0.05_C1.D9-UP.subset.GOseq.enriched
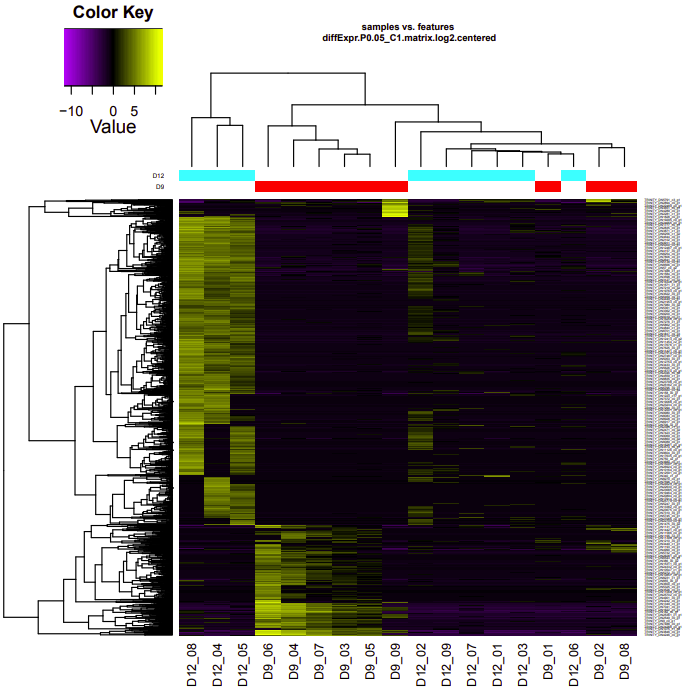
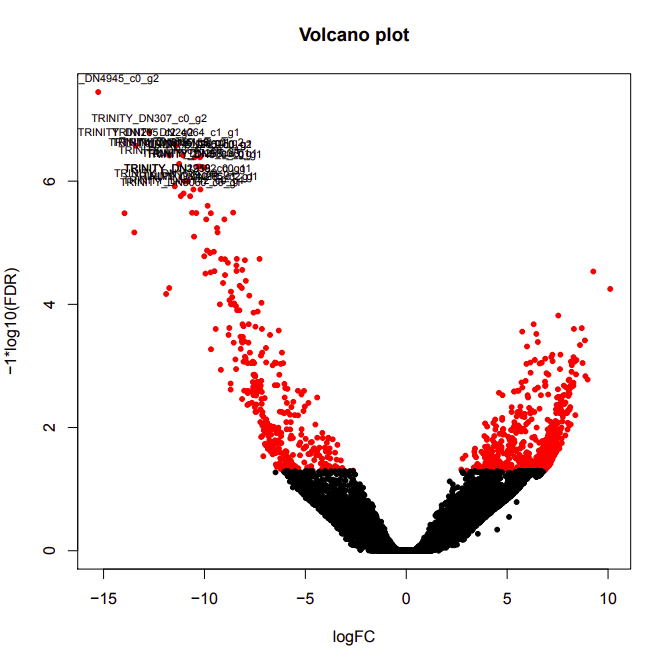
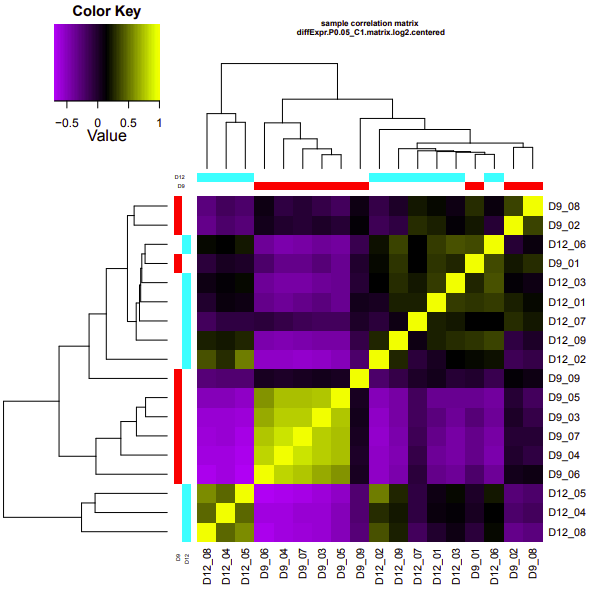
D9-D26
Up-regulated genes:
salmon.gene.counts.matrix.D26_vs_D9.edgeR.DE_results.P0.05_C1.D9-UP.subset
salmon.gene.counts.matrix.D26_vs_D9.edgeR.DE_results.P0.05_C1.D26-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.D26_vs_D9.edgeR.DE_results.P0.05_C1.D9-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.D26_vs_D9.edgeR.DE_results.P0.05_C1.D26-UP.subset.GOseq.enriched
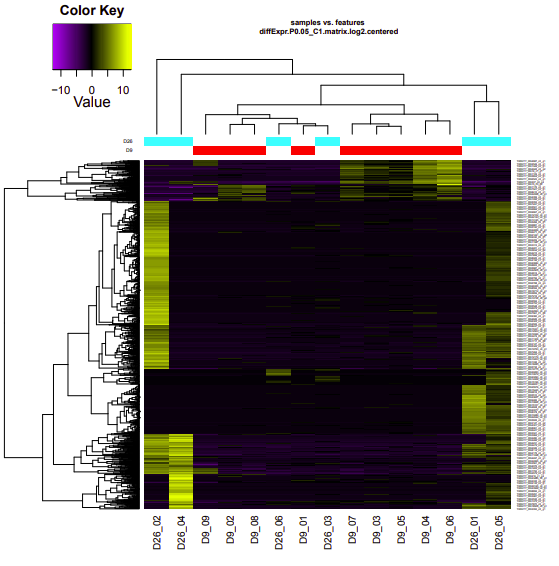
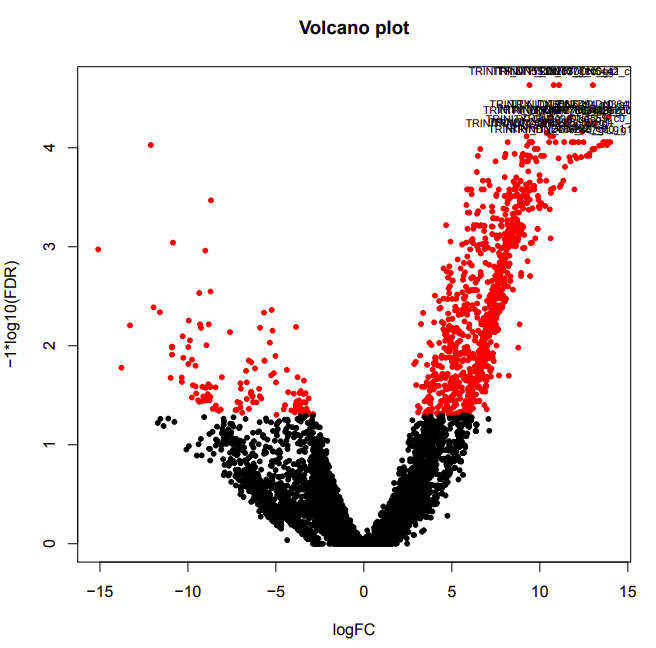
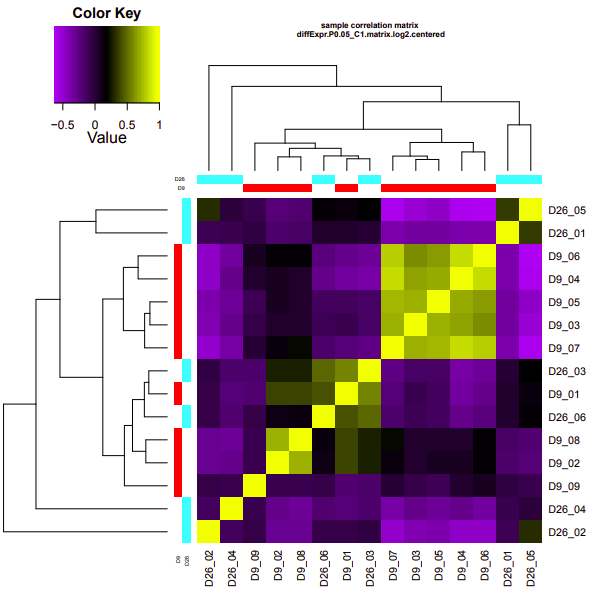
D12-D26
Up-regulated genes:
salmon.gene.counts.matrix.D12_vs_D26.edgeR.DE_results.P0.05_C1.D26-UP.subset
salmon.gene.counts.matrix.D12_vs_D26.edgeR.DE_results.P0.05_C1.D12-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.D12_vs_D26.edgeR.DE_results.P0.05_C1.D26-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.D12_vs_D26.edgeR.DE_results.P0.05_C1.D12-UP.subset.GOseq.enriched
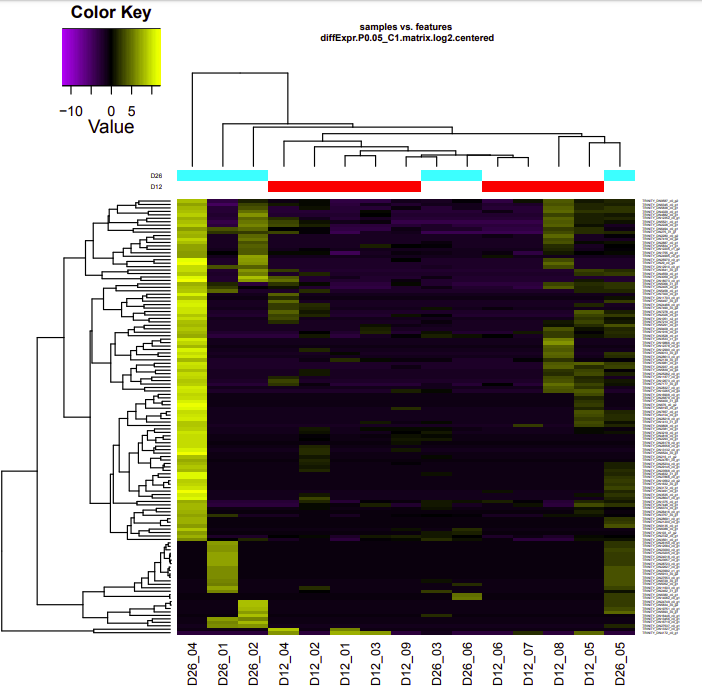
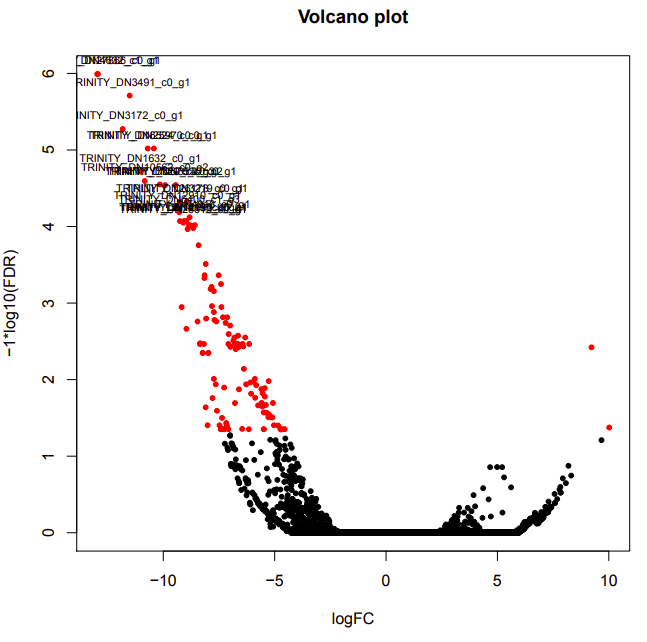
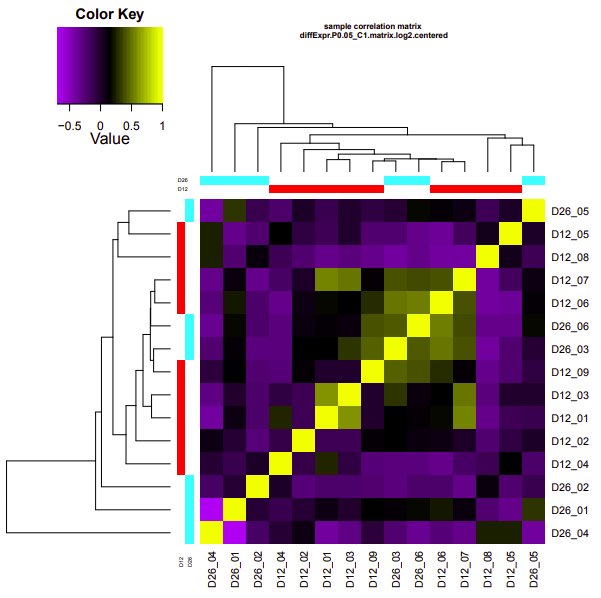
ambient-cold
Up-regulated genes:
salmon.gene.counts.matrix.ambient_vs_cold.edgeR.DE_results.P0.05_C1.cold-UP.subset
salmon.gene.counts.matrix.ambient_vs_cold.edgeR.DE_results.P0.05_C1.ambient-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.ambient_vs_cold.edgeR.DE_results.P0.05_C1.cold-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.ambient_vs_cold.edgeR.DE_results.P0.05_C1.ambient-UP.subset.GOseq.enriched
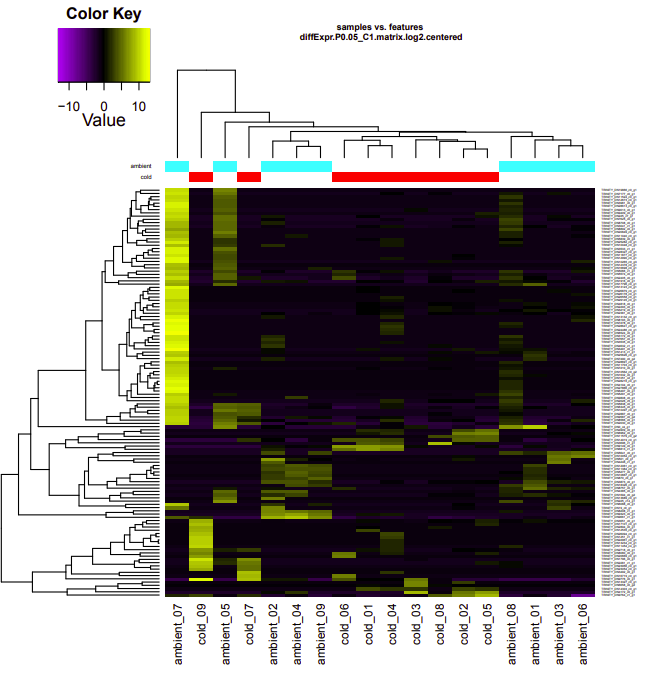
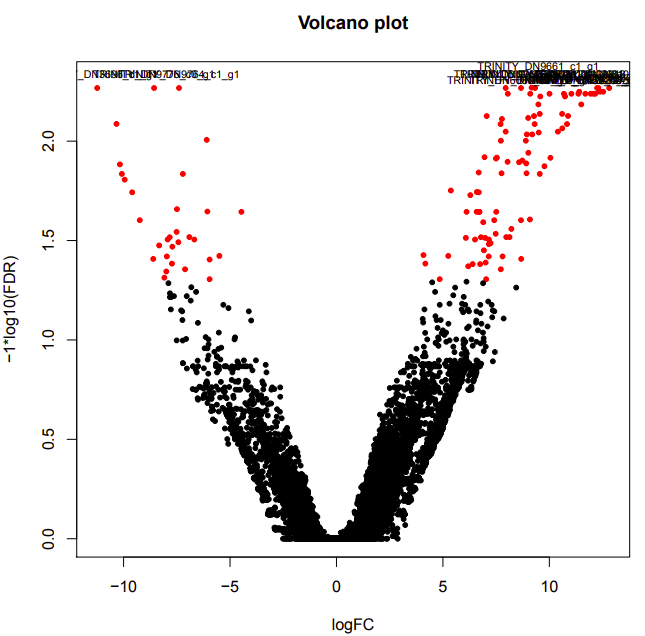
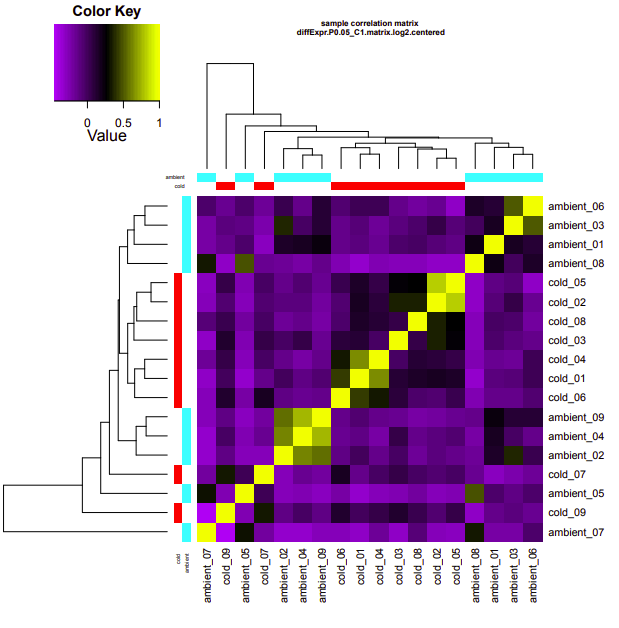
ambient-warm
Up-regulated genes:
salmon.gene.counts.matrix.ambient_vs_warm.edgeR.DE_results.P0.05_C1.ambient-UP.subset
salmon.gene.counts.matrix.ambient_vs_warm.edgeR.DE_results.P0.05_C1.warm-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.ambient_vs_warm.edgeR.DE_results.P0.05_C1.warm-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.ambient_vs_warm.edgeR.DE_results.P0.05_C1.ambient-UP.subset.GOseq.enriched
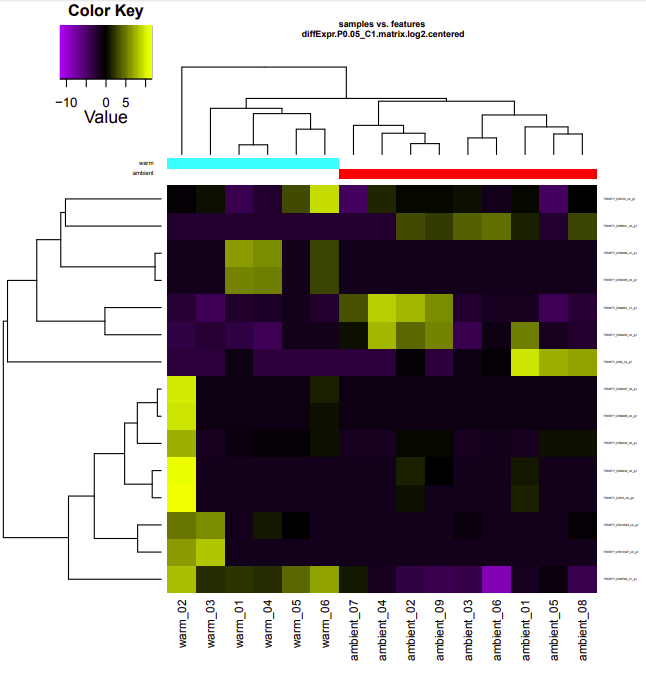
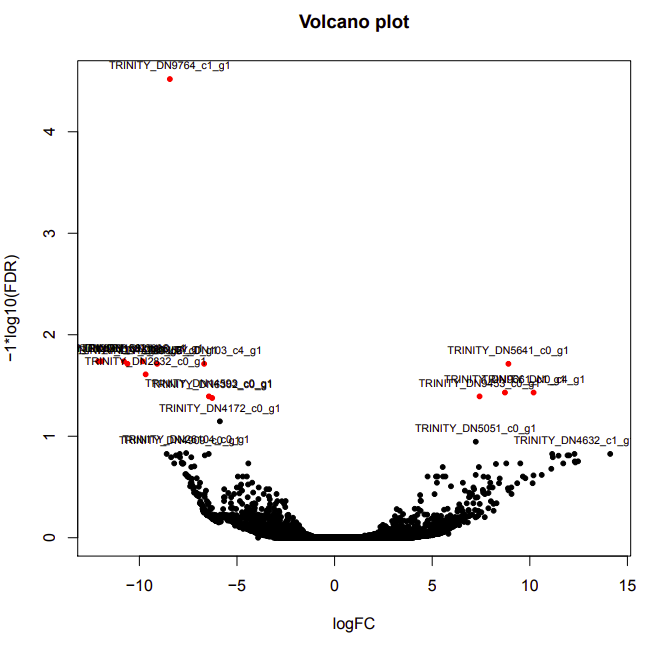
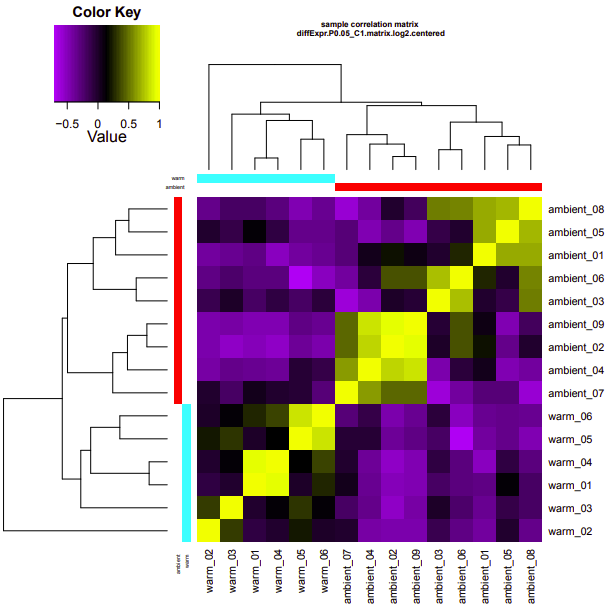
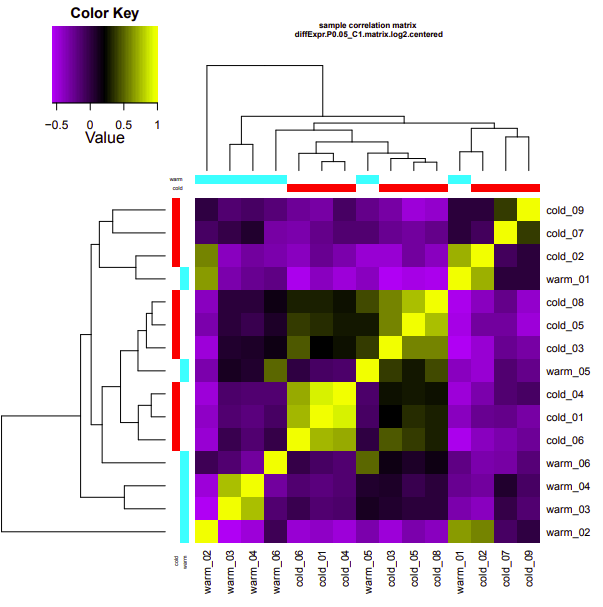
cold-warm
Up-regulated genes:
salmon.gene.counts.matrix.cold_vs_warm.edgeR.DE_results.P0.05_C1.warm-UP.subset
salmon.gene.counts.matrix.cold_vs_warm.edgeR.DE_results.P0.05_C1.cold-UP.subset
Enriched GO terms:
salmon.gene.counts.matrix.cold_vs_warm.edgeR.DE_results.P0.05_C1.warm-UP.subset.GOseq.enriched
salmon.gene.counts.matrix.cold_vs_warm.edgeR.DE_results.P0.05_C1.cold-UP.subset.GOseq.enriched
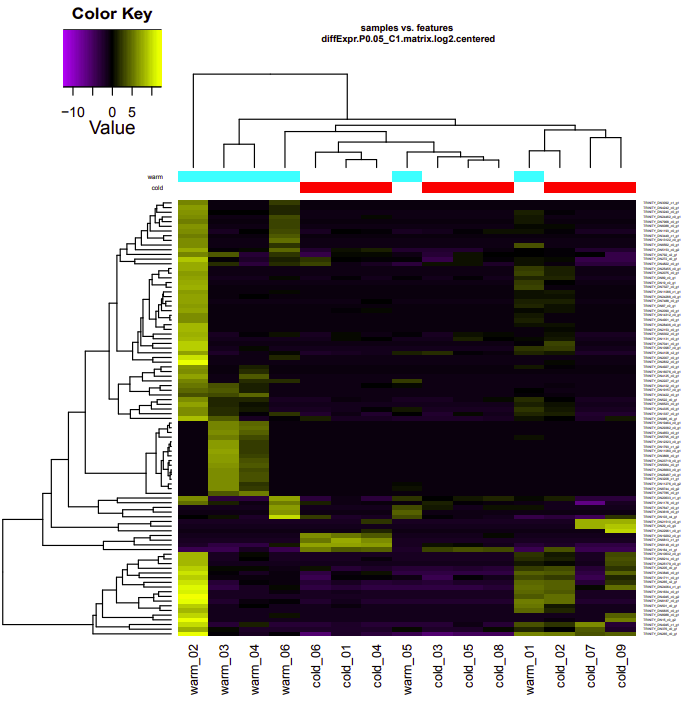
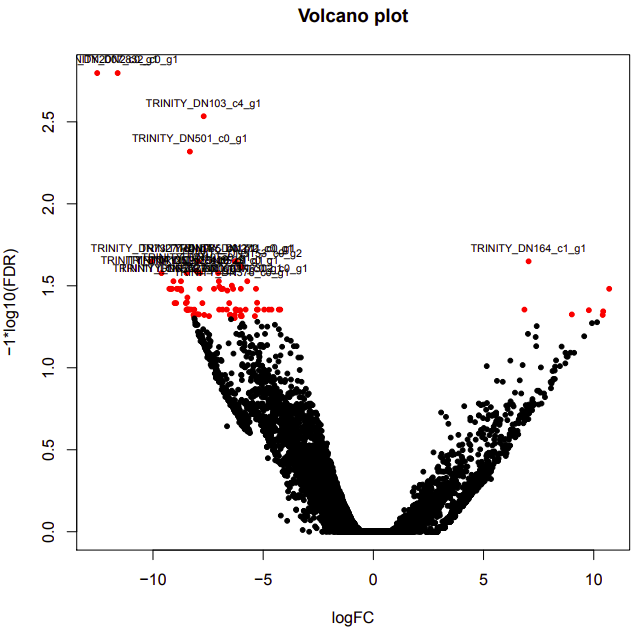
infected-uninfected
Up-regulated genes:
salmon.gene.counts.matrix.infected_vs_uninfected.edgeR.DE_results.P0.05_C1.infected-UP.subset
salmon.gene.counts.matrix.infected_vs_uninfected.edgeR.DE_results.P0.05_C1.uninfected-UP.subset
Enriched GO terms: