Steven asked that I obtain relative expression values for various geoduck tissues (GitHub Issue). So, I decided to use this as an opportunity to try to use a Nextflow pipeline. There’s an RNAseq pipeline, NF-Core RNAseq which I decided to use. The pipeline appears to be ridiculously thorough (e.g. trims, removes gDNA/rRNA contamination, allows for multiple aligners to be used, quantifies/visualizes feature assignments by reads, performs differential gene expression analysis and visualization), all in one package. Sounds great, but I did have some initial problems getting things up and running. Overall, getting things set up to actually run took longer than the actual pipeline run! Oh well, it’s a learning process, so that’s not totally unexpected.
For this pipeline run, I made some modifications to the genome GFF input file used. First, I attempted to create a “gene_biotype” description for the pipeline to use to get some visualizations of read assignments to different components of the genome. I did that in the following fashion:
# Copies header to new GFF
awk 'NR < 4 {print $0}' Panopea-generosa-v1.0.gff > Panopea-generosa-v1.0_biotype.gff
# Adds "gene_biotype" to end of line that matches feature field ($3)
awk 'NR > 3 {print $0";gene_biotype="$3}' Panopea-generosa-v1.0.gff >> Panopea-generosa-v1.0_biotype.gffThen, modified it further to convert tRNA strand to + instead of . in order to avoid RSEM errors regarding strand info and removed RNAmmer features to also avoid RSEM strand errors.
# Converts strand field ($7) to `+` instead of `.`.
# Works just on tRNA entries
awk '$2 == "GenSAS_5d82b316cd298-trnascan" {$7="+"}1' Panopea-generosa-v1.0.a4_biotype.gff > Panopea-generosa-v1.0.a4_biotype-trna_strand_converted.gff
# Prints all lines which are not rRNA
awk '$2 != "RNAmmer-1.2"' Panopea-generosa-v1.0.a4_biotype-trna_strand_converted.gff > Panopea-generosa-v1.0.a4_biotype-trna_strand_converted-no_RNAmmer.gffThen, this was all run on Mox.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=20220323-pgen-nextflow_rnaseq-tissues
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=17-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20220323-pgen-nextflow_rnaseq-tissues
# Script to run Nextflow NF Core RNAseq pipeline for RNAseq analysis of P.generosa, per this GitHub Issue:
# https://github.com/RobertsLab/resources/issues/1423
# See variable assignments below for input files used: genome, GFF, transcriptome
# List of input FastQs will be generated during run in: sample_sheet-"${SLURM_JOB_ID}".csv
# Custom config file for maximum memory and CPU thread setttings
# Outputs explanations are here: https://nf-co.re/rnaseq/3.6/docs/output/
# Input parameter explanations are here: https://nf-co.re/rnaseq/3.6/parameters
###################################################################################
# These variables need to be set by user
## Assign Variables
## PROGRAMS ##
# NF Core RNAseq workflow directory
nf_core_rnaseq="/gscratch/srlab/programs/nf-core-rnaseq-3.6/workflow"
# NF Core RNAseq custom config file
nf_core_rnaseq_config=/gscratch/srlab/programs/nf-core-rnaseq-3.6/configs/conf/base-srlab_500GB_node.config
## FILES AND DIRECTORIES ##
# Wordking directory
wd=$(pwd)
# RNAseq FastQs directory
reads_dir=/gscratch/srlab/sam/data/P_generosa/RNAseq
# Genome FastA
genome_fasta=/gscratch/srlab/sam/data/P_generosa/genomes/Panopea-generosa-v1.0.fa
# Genome GFF3
# This was manually modified by me to add gene_biotype to end of each entry.
# Improves NF-Core RNAseq pipeline analysis/visualiztion to have this info present.
genome_gff=/gscratch/srlab/sam/data/P_generosa/genomes/Panopea-generosa-v1.0.a4_biotype-trna_strand_converted-no_RNAmmer.gff
## INITIALIZE ARRAYS ##
# Leave empty!!
R1_array=()
R2_array=()
R1_uncompressed_array=()
R2_uncompressed_array=()
###################################################################################
# Exit script if a command fails
set -e
# Load Anaconda
# Uknown why this is needed, but Anaconda will not run if this line is not included.
. "/gscratch/srlab/programs/anaconda3/etc/profile.d/conda.sh"
# Activate NF-core conda environment
conda activate nf-core_env
# Load Singularity Mox module for NF Core/Nextflow
module load singularity
# NF Core RNAseq sample sheet header
sample_sheet_header="sample,fastq_1,fastq_2,strandedness"
printf "%s\n" "${sample_sheet_header}" >> sample_sheet-"${SLURM_JOB_ID}".csv
# Create array of original uncompressed fastq R1 files
# Set filename pattern
R1_uncompressed_array=("${reads_dir}"/*_1.fastq)
# Create array of original uncompressed fastq R2 files
# Set filename pattern
R2_uncompressed_array=("${reads_dir}"/*_2.fastq)
# Check array size to confirm it has all expected samples
# Exit if mismatch
if [[ "${#R1_uncompressed_array[@]}" != "${#R2_uncompressed_array[@]}" ]]
then
echo ""
echo "Uncompressed array sizes don't match."
echo "Confirm all expected FastQs are present in ${reads_dir}"
echo ""
exit
fi
# Create list of original uncompressed fastq files
## Uses parameter substitution to strip leading path from filename
for fastq in "${!R1_uncompressed_array[@]}"
do
# Strip leading path
no_path=$(echo "${R1_uncompressed_array[${fastq}]##*/}")
# Grab SRA name
sra=$(echo "${no_path}" | awk -F "_" '{print $1}')
# Only gzip matching FastQs
# Only generate MD5 checksums for matching FastQs
if [[ "${sra}" == "SRR12218868" ]] \
|| [[ "${sra}" == "SRR12218869" ]] \
|| [[ "${sra}" == "SRR12226692" ]] \
|| [[ "${sra}" == "SRR12218870" ]] \
|| [[ "${sra}" == "SRR12226693" ]] \
|| [[ "${sra}" == "SRR12207404" ]] \
|| [[ "${sra}" == "SRR12207405" ]] \
|| [[ "${sra}" == "SRR12227930" ]] \
|| [[ "${sra}" == "SRR12207406" ]] \
|| [[ "${sra}" == "SRR12207407" ]] \
|| [[ "${sra}" == "SRR12227931" ]] \
|| [[ "${sra}" == "SRR12212519" ]] \
|| [[ "${sra}" == "SRR12227929" ]] \
|| [[ "${sra}" == "SRR8788211" ]]
then
echo ""
echo "Generating MD5 checksums for ${R1_uncompressed_array[${fastq}]} and ${R2_uncompressed_array[${fastq}]}."
echo ""
# Generate MD5 checksums of uncompressed FastQs
{
md5sum "${R1_uncompressed_array[${fastq}]}"
md5sum "${R2_uncompressed_array[${fastq}]}"
} >> uncompressed_fastqs-"${SLURM_JOB_ID}".md5
# Gzip FastQs; NF Core RNAseq requires gzipped FastQs as inputs
echo "Compressing FastQ files."
if [ ! -f "${R1_uncompressed_array[${fastq}]}.gz" ]
then
gzip --keep "${R1_uncompressed_array[${fastq}]}"
gzip --keep "${R2_uncompressed_array[${fastq}]}"
else
echo "${R1_uncompressed_array[${fastq}]}.gz already exists. Skipping."
fi
echo ""
fi
done
# Create array of fastq R1 files
# Set filename pattern
R1_array=("${reads_dir}"/*_1.fastq.gz)
# Create array of fastq R2 files
# Set filename pattern
R2_array=("${reads_dir}"/*_2.fastq.gz)
# Check array sizes to confirm they are same size
# Exit if mismatch
if [[ "${#R1_array[@]}" != "${#R2_array[@]}" ]]
then
echo ""
echo "Read1 and Read2 compressed FastQ array sizes don't match."
echo "Confirm all expected compressed FastQs are present in ${reads_dir}"
echo ""
exit
fi
# Create list of fastq files used in analysis
## Uses parameter substitution to strip leading path from filename
for fastq in "${!R1_array[@]}"
do
echo ""
echo "Generating MD5 checksums for ${R1_array[${fastq}]} and ${R2_array[${fastq}]}."
echo ""
# Generate MD5 checksums for compressed FastQs used in NF Core RNAseq analysis
{
md5sum "${R1_array[${fastq}]}"
md5sum "${R2_array[${fastq}]}"
} >> input_fastqs-"${SLURM_JOB_ID}".md5
# Strip leading path
no_path=$(echo "${R1_array[${fastq}]##*/}")
# Grab SRA name
sra=$(echo "${no_path}" | awk -F "_" '{print $1}')
# Set tissue type
if [[ "${sra}" == "SRR12218868" ]]
then
tissue="heart"
# Add to NF Core RNAseq sample sheet
printf "%s,%s,%s,%s\n" "${tissue}" "${R1_array[${fastq}]}" "${R2_array[${fastq}]}" "reverse" \
>> sample_sheet-"${SLURM_JOB_ID}".csv
elif [[ "${sra}" == "SRR12218869" ]] \
|| [[ "${sra}" == "SRR12226692" ]]
then
tissue="gonad"
# Add to NF Core RNAseq sample sheet
printf "%s,%s,%s,%s\n" "${tissue}" "${R1_array[${fastq}]}" "${R2_array[${fastq}]}" "reverse" \
>> sample_sheet-"${SLURM_JOB_ID}".csv
elif [[ "${sra}" == "SRR12218870" ]] \
|| [[ "${sra}" == "SRR12226693" ]]
then
tissue="ctenidia"
# Add to NF Core RNAseq sample sheet
printf "%s,%s,%s,%s\n" "${tissue}" "${R1_array[${fastq}]}" "${R2_array[${fastq}]}" "reverse" \
>> sample_sheet-"${SLURM_JOB_ID}".csv
elif [[ "${sra}" == "SRR12207404" ]] \
|| [[ "${sra}" == "SRR12207405" ]] \
|| [[ "${sra}" == "SRR12227930" ]] \
|| [[ "${sra}" == "SRR12207406" ]] \
|| [[ "${sra}" == "SRR12207407" ]] \
|| [[ "${sra}" == "SRR12227931" ]]
then
tissue="juvenile"
# Add to NF Core RNAseq sample sheet
printf "%s,%s,%s,%s\n" "${tissue}" "${R1_array[${fastq}]}" "${R2_array[${fastq}]}" "reverse" \
>> sample_sheet-"${SLURM_JOB_ID}".csv
elif [[ "${sra}" == "SRR12212519" ]] \
|| [[ "${sra}" == "SRR12227929" ]] \
|| [[ "${sra}" == "SRR8788211" ]]
then
tissue="larvae"
# Add to NF Core RNAseq sample sheet
printf "%s,%s,%s,%s\n" "${tissue}" "${R1_array[${fastq}]}" "${R2_array[${fastq}]}" "reverse" \
>> sample_sheet-"${SLURM_JOB_ID}".csv
fi
done
echo "Beginning NF-Core RNAseq pipeline..."
echo ""
# Run NF Core RNAseq workflow
nextflow run ${nf_core_rnaseq} \
-profile singularity \
-c ${nf_core_rnaseq_config} \
--input sample_sheet-"${SLURM_JOB_ID}".csv \
--outdir ${wd} \
--multiqc_title "20220321-pgen-nextflow_rnaseq-tissues-${SLURM_JOB_ID}" \
--fasta ${genome_fasta} \
--gff ${genome_gff} \
--save_reference \
--gtf_extra_attributes gene_name \
--gtf_group_features gene_id \
--featurecounts_group_type gene_biotype \
--featurecounts_feature_type exon \
--trim_nextseq 20 \
--save_trimmed \
--aligner star_salmon \
--pseudo_aligner salmon \
--min_mapped_reads 5 \
--save_align_intermeds \
--rseqc_modules bam_stat,\
inner_distance,\
infer_experiment,\
junction_annotation,\
junction_saturation,\
read_distribution,\
read_duplication
##############################################################
# Copy config file for later reference, if needed
cp "${nf_core_rnaseq_config}" .
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
echo "Finished logging system PATH"RESULTS
Runtime was surprisingly fast, at just a bit over 2.3 days…

There is a ton of data here to unpack, so I’ll just link to some of the most useful files.
Refer to the NF-Core/RNAseq “Output docs” for all the things generated, as well as a thorough explanation of the MultiQC Report:
Also, the NF-Core/RNAseq pipeline provides a nice progress report to show you which options are running/completed. This screenshot is from after the pipeline finished successfully:
Output folder:
20220323-pgen-nextflow_rnaseq-tissues/
Pipeline MultiQC Report (HTML - opens interactive report in browser):
One interesting thing I noticed in this report is the disproportionate number of reads in gonad samples mapping to Scaffold_08 in the genome. This suggests that there is a cluster of genes invovled in reproduction on Scaffold_08.
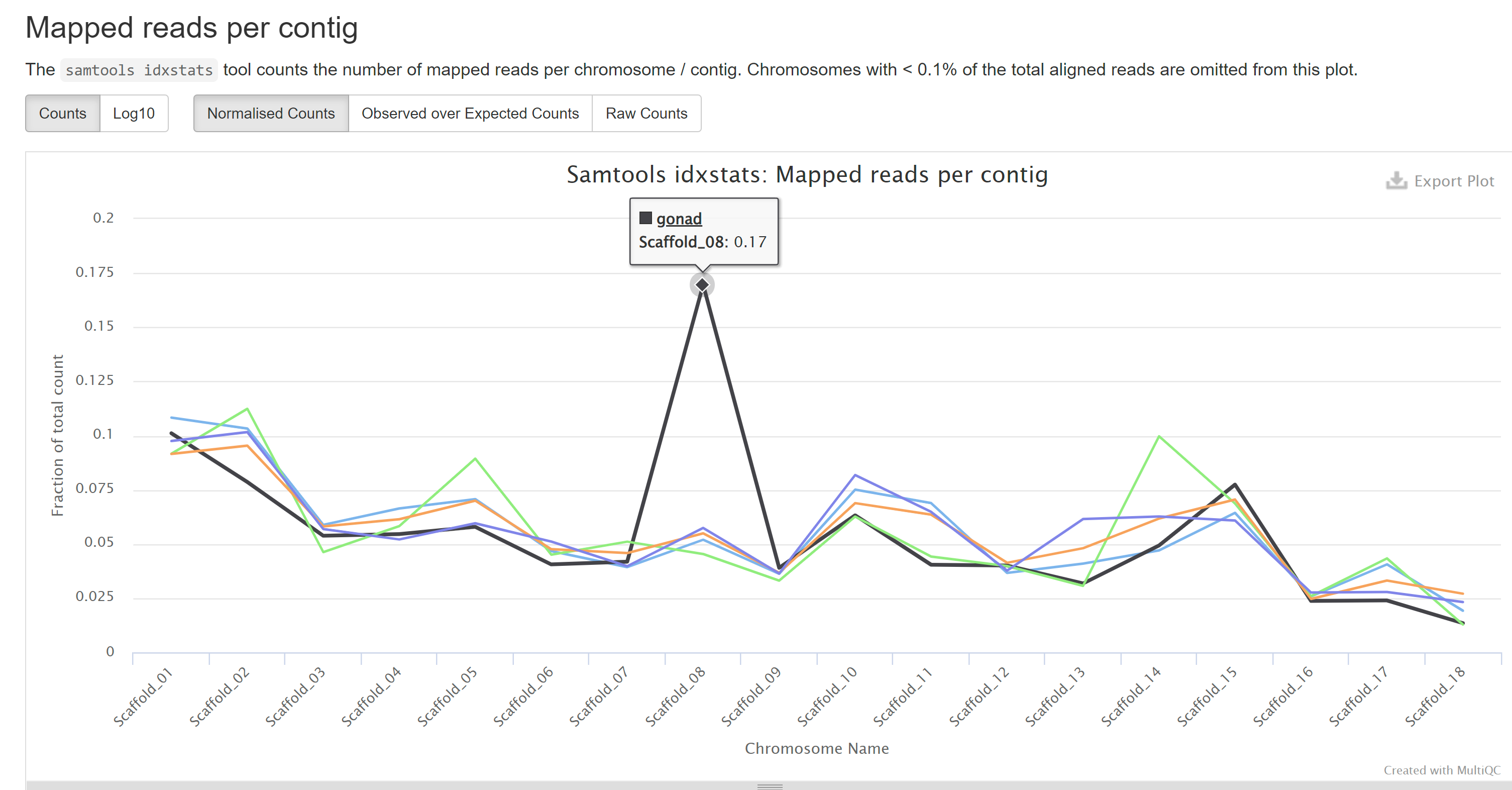
Histogram showing normalized read counts of samples mapping to each of the 18 P.generosa genome scaffolds. A noticable spike in the black line (gonad) occurs in Scaffold 08. List of compressed FastQ files and MD5 checksums used (text):
List of original, uncompressed FastQ files and MD5 checksums (text):
Sample sheet used as input to NF-Core RNAseq pipeline (CSV):
Config file used as input to NF-Core RNAseq pipeline (text). Mostly used to specify CPUs, RAM, and runtimes:
STAR alignments (BAM):
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/ctenidia.markdup.sorted.bam (4.2G)
- MD5:
b5a8d02851184dbbf1155cfb2a7b4800
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/gonad.markdup.sorted.bam (3.8G)
- MD5:
fe2e49e4e81de935eb23c5f095e167f1
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/heart.markdup.sorted.bam (7.9G)
- MD5:
3b621ed144399b27a5880416a9d9e98b
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/juvenile.markdup.sorted.bam (26G)
- MD5:
f191d39dee3ea29964ee69861c48e01a
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/larvae.markdup.sorted.bam (5.0G)
- MD5:
15a215edcde2ae73c46940a4680e9bd4
- MD5:
Ballgown table folders:
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/stringtie/ctenidia.ballgown/ (31M)
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/stringtie/gonad.ballgown/ (31M)
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/stringtie/heart.ballgown/ (31M)
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/stringtie/juvenile.ballgown/ (32M)
20220323-pgen-nextflow_rnaseq-tissues/star_salmon/stringtie/larvae.ballgown/ (31M)
STAR/Salmon TPM (text):
Salmon ONLY
quant.sffiles (text) can be found in tissue subdirectories:STAR and Salmon genome index files in respective subdirectories:
Trimmed FastQs:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/ctenidia_1_val_1.fq.gz (4.3G)
- MD5:
fb5683c9734fd90e95f8b09292626f74
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/ctenidia_2_val_2.fq.gz (4.4G)
- MD5:
a124c1ce517cd106979a32d0a9957742
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/gonad_1_val_1.fq.gz (4.7G)
- MD5:
e999349a51d70f6f21384126b42bbe5e
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/gonad_2_val_2.fq.gz (4.9G)
- MD5:
f70ac9c1170120f67e54cd523e8284eb
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/heart_1_val_1.fq.gz (8.0G)
- MD5:
d5d930b6f92005f3acdb8105736d2219
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/heart_2_val_2.fq.gz (7.8G)
- MD5:
0068df5bf32d18c9a940a4558645021e
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/juvenile_1_val_1.fq.gz (26G)
- MD5:
9f74db3f73fc140e009c470010c5b5b2
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/juvenile_2_val_2.fq.gz (26G)
- MD5:
453b846f816e227f386481b8613aee86
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/larvae_1_val_1.fq.gz (5.3G)
- MD5:
70ff0f167449b7952362eba1c6b8ef18
- MD5:
20220323-pgen-nextflow_rnaseq-tissues/trimgalore/larvae_2_val_2.fq.gz (5.5G)
- MD5:
9f4b1080d956cfab03b94bba403ee81d
- MD5: