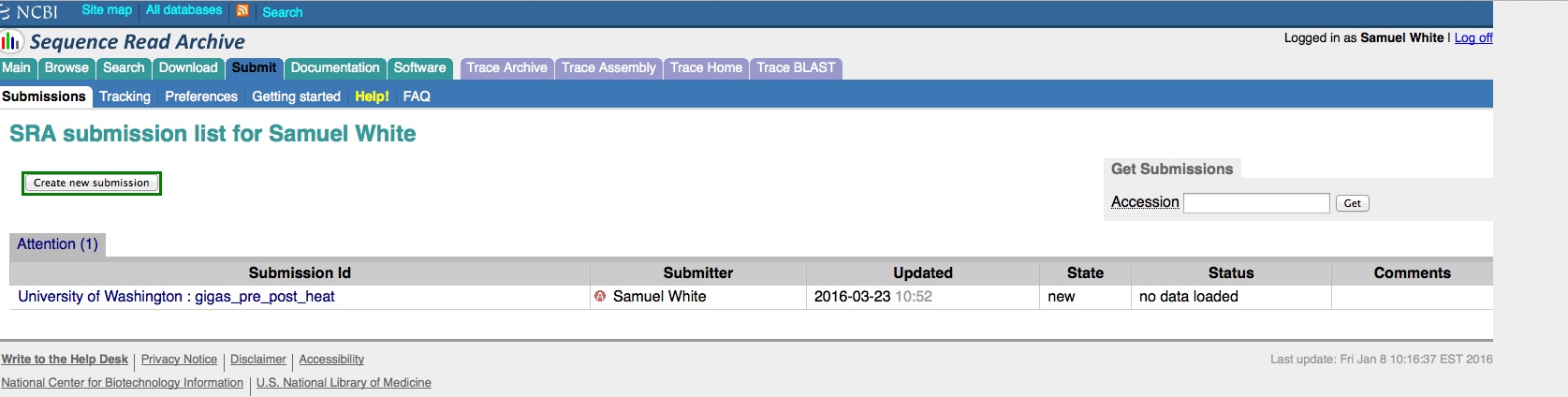
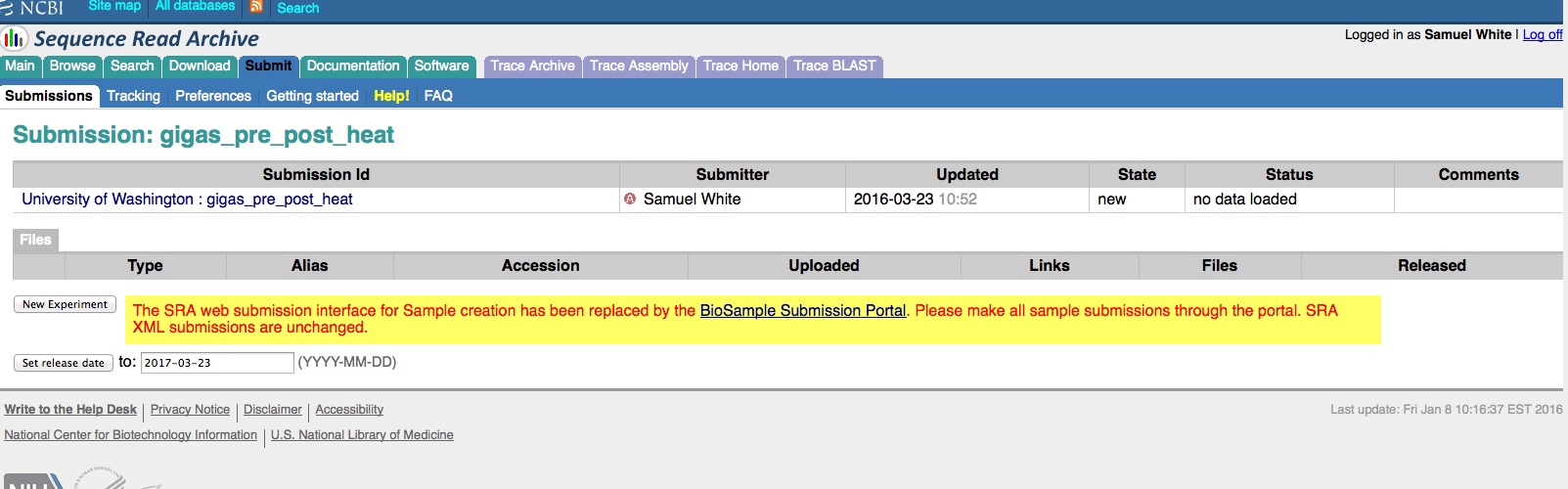
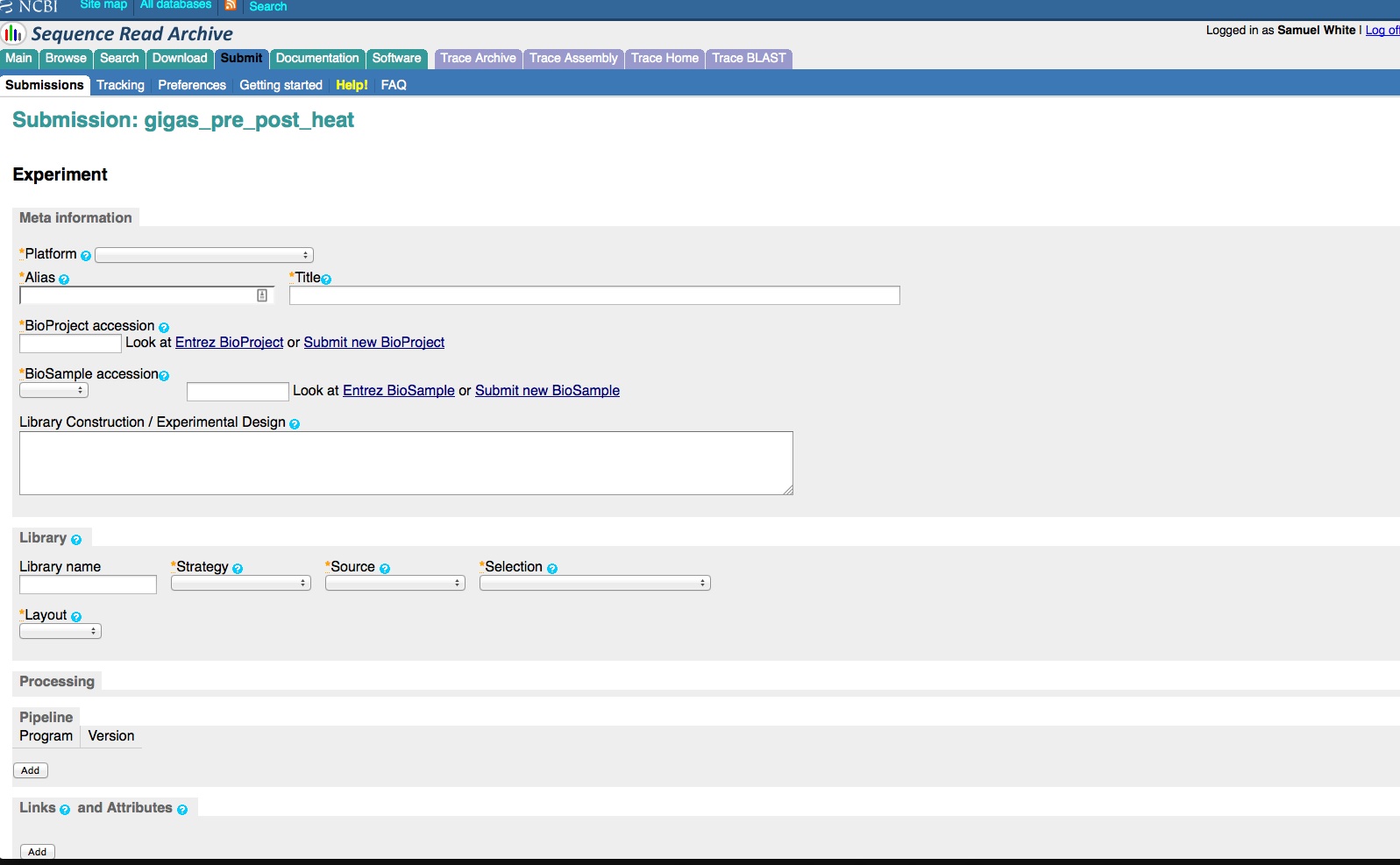
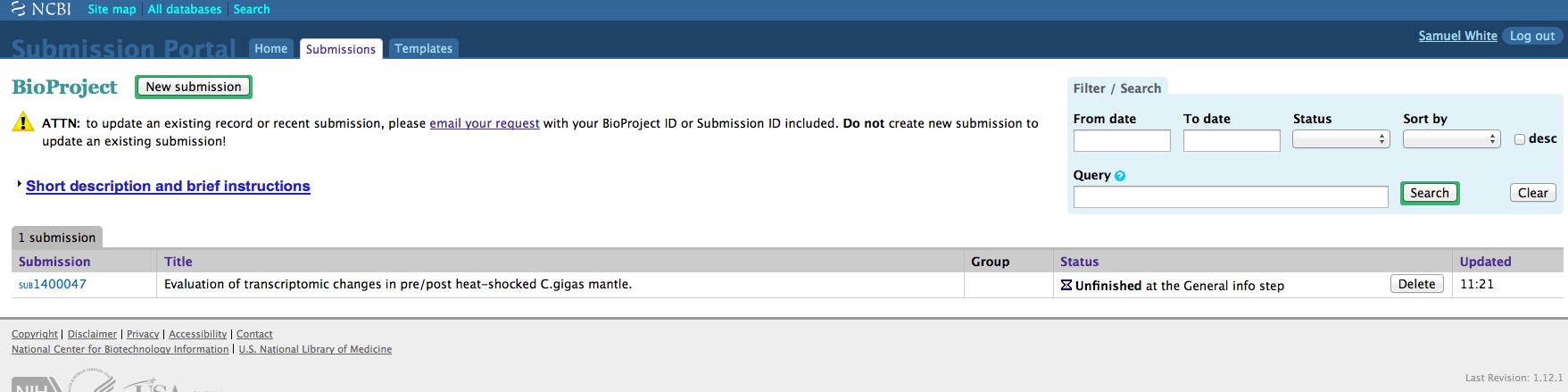
We have an enormous backlog of high-throughput sequencing files (641 FASTQ files, to be exact) that we need/want to get added to the NCBI Sequence Read Archive (SRA).
This post provides a brief summary of what’s involved in the process (mostly via screen shots) and attempts to identify the various pitfalls/pains that I’ve already stumbled through trying to get a set of six FASTQ files submitted properly.
OVERALL - It’s horrible and tedious.
Important things to note:
Once any of the three required components for SRA submission have been created (SRA, BioProject, and BioSamples), they can no longer be edited/deleted by the user! Understandable if they’ve already been publicly released, but if they’re still in pre-public release status, I think the user should be able to make changes as they see fit. As it currently stands, the user has to email the help desk at SRA and/or BioProjects to make any changes.
Extremely difficult to figure out which information will show up (and where it will show up) in the final, formatted SRA record - no guide to this that I could find. Thus, if you screw it up, it’s a major, major hassle to try to change anything.
When creating a “Run” (within an “Experiment”, within your SRA submission), only include sequencing files that provide the same data (e.g. if you have multiple sequence files, each generated from different individuals/samples, then you need to create a separate “Experiment” and “Run” for each of those files - otherwise, all files uploaded to a “Run” are combined into a single SRA file that loses any distinguishing info from the separate sequencing files).
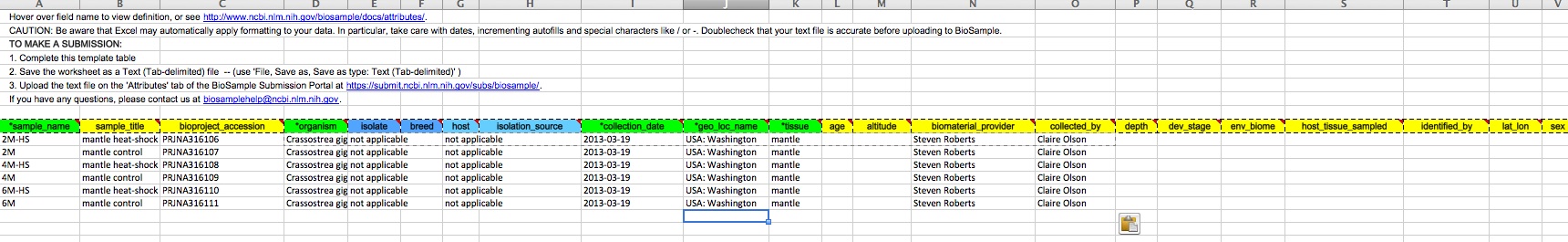
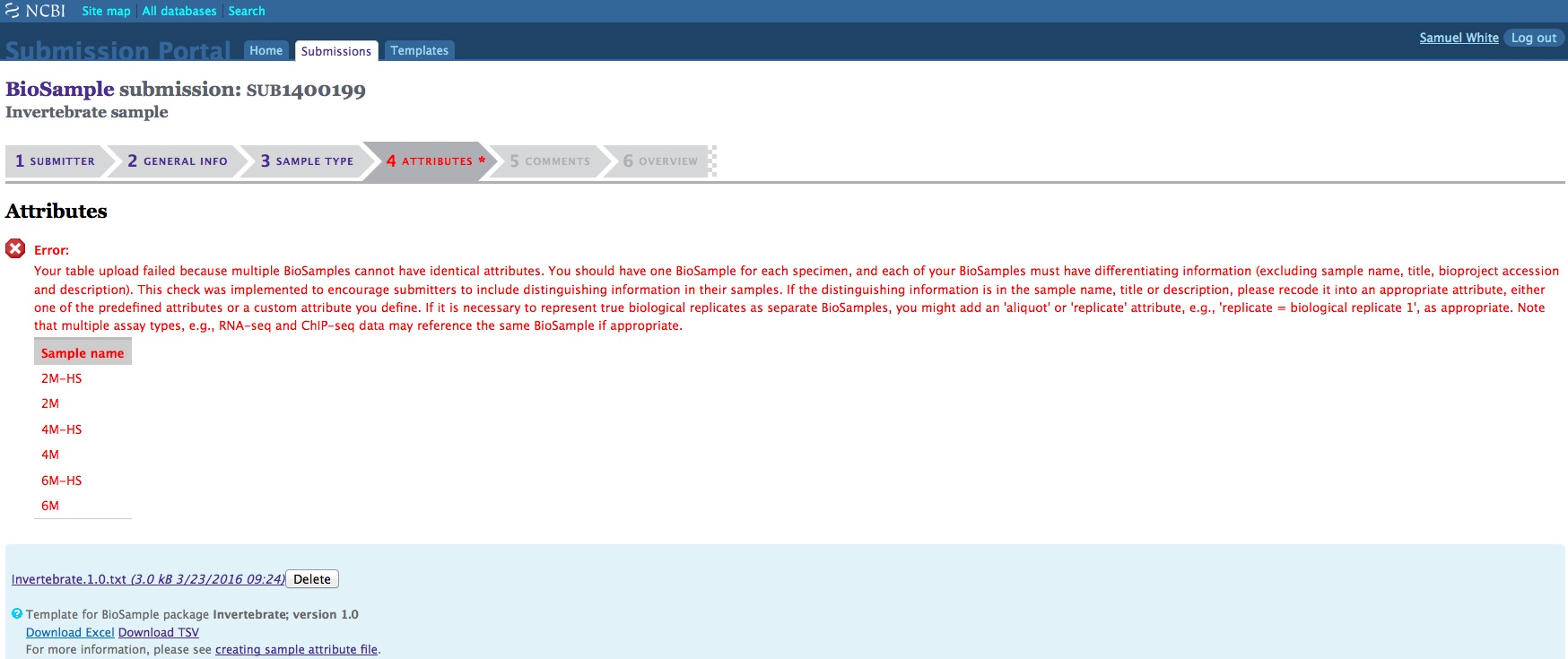
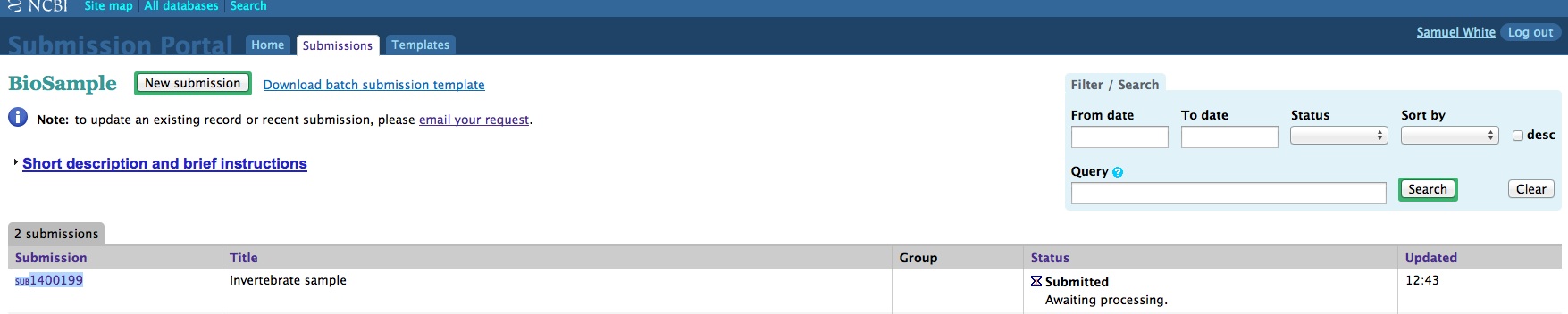
When creating a batch submission for BioSamples, there’s no way to set a Title attribute. This means all of your submissions (in my case) will have all have a title of “Invertebrate sample”. Considering that I will likely end up with dozens of BioSamples, that means there’s no easy way to distinguish them from each other without some extra clicking and poking around.
Here’s the best way to proceed:
Create a BioProject. This will sit at the top of the hierarchy in the SRA submission and will be displayed as the STUDY associated with the SRA.
Create BioSample(s). This will be the next level of the hierarchy in the SRA submission and will be displayed as SAMPLE. This only shows up in the SRA when you create a new “Experiment”
Create SRA. This will end up encompassing any BioProject(s) and BioSample(s) that you need to include to describe the sequencing files you’re submitting to the SRA.
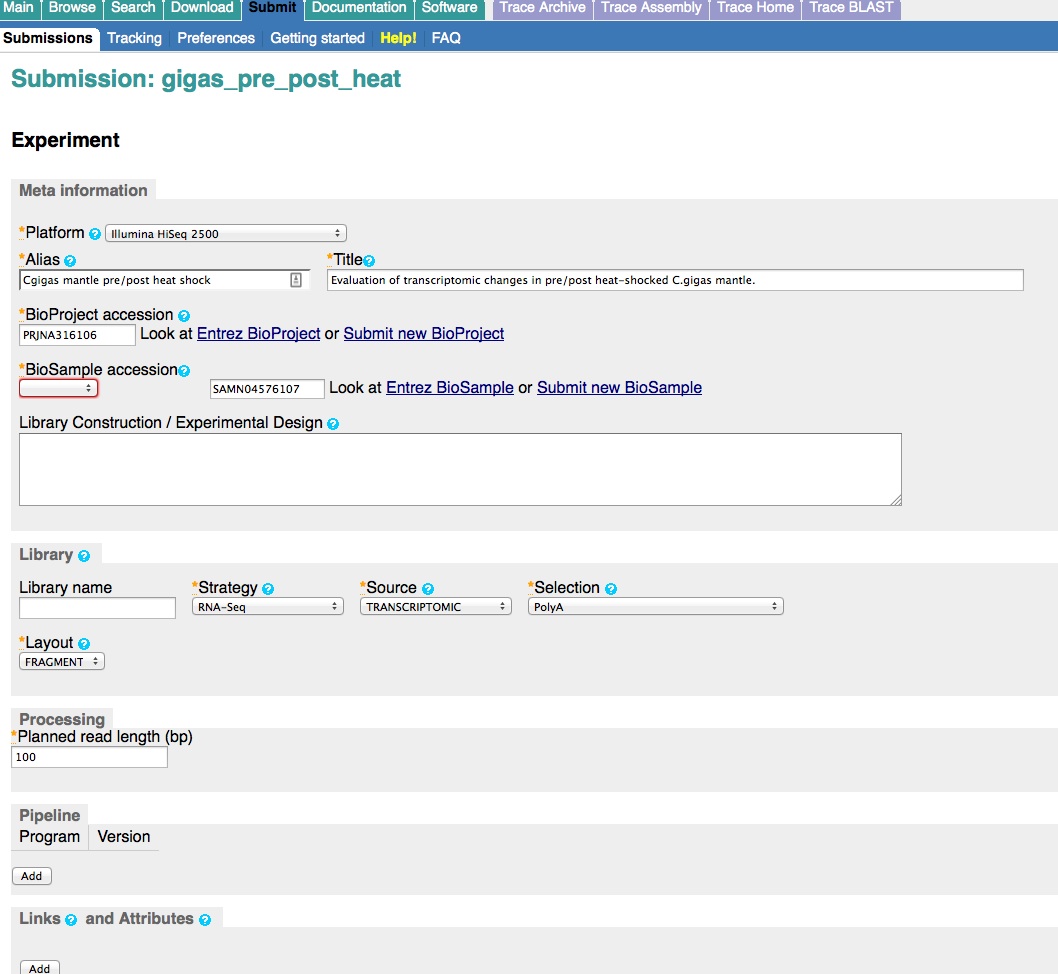
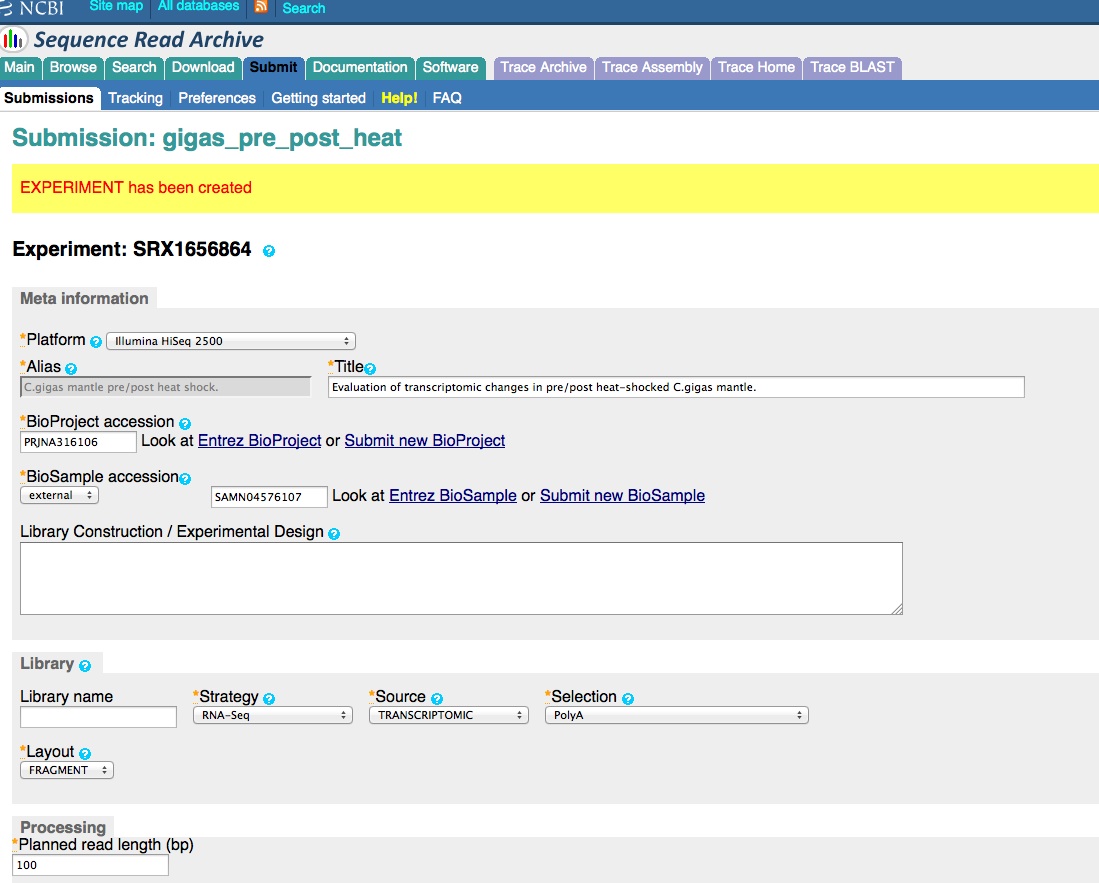
Create an Experiment.
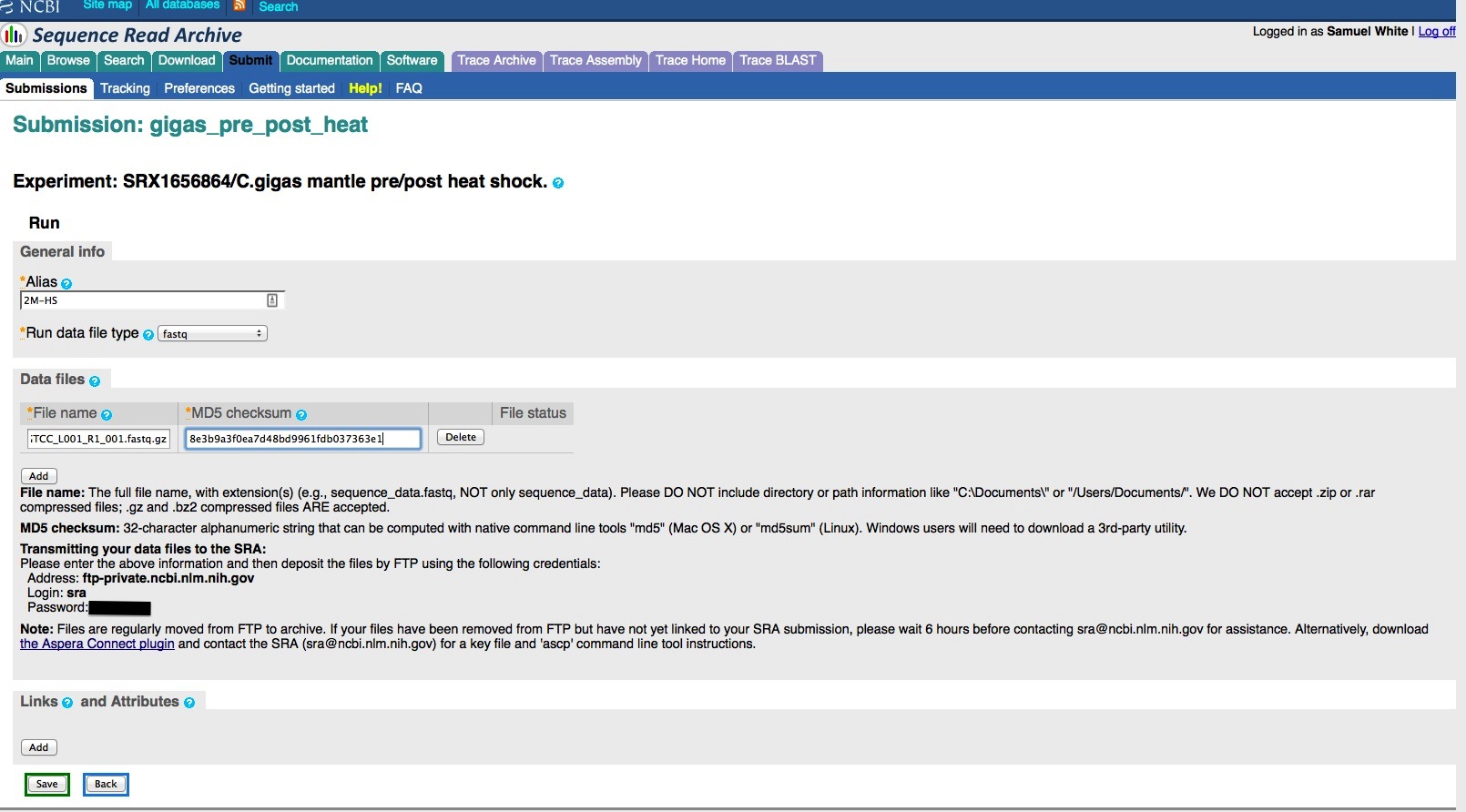
Create a Run. This option is available once you’ve saved your experiment. This is where you provide your sequencing filename and associated MD5 checksum. This will also provide you with the login info to upload your sequencing files via FTP to NCBI servers. You can associate multiple sequencing files within a single run. This should be done if your sequencing files all provide data for the BioSample you selected. However, if you have sequencing files that are associated with different BioSamples, then you need to create an individual Experiment (and Run) for each BioSample!
Here are some links that might come in handy (although, none are that great)…
SRA Getting Started (helpful):
SRA Metadata Overview (this is helpful):
SRA Submission Quick Start Guide (this is useful!):
FTP Upload Instructions:
User UN-friendly SRA Guide:
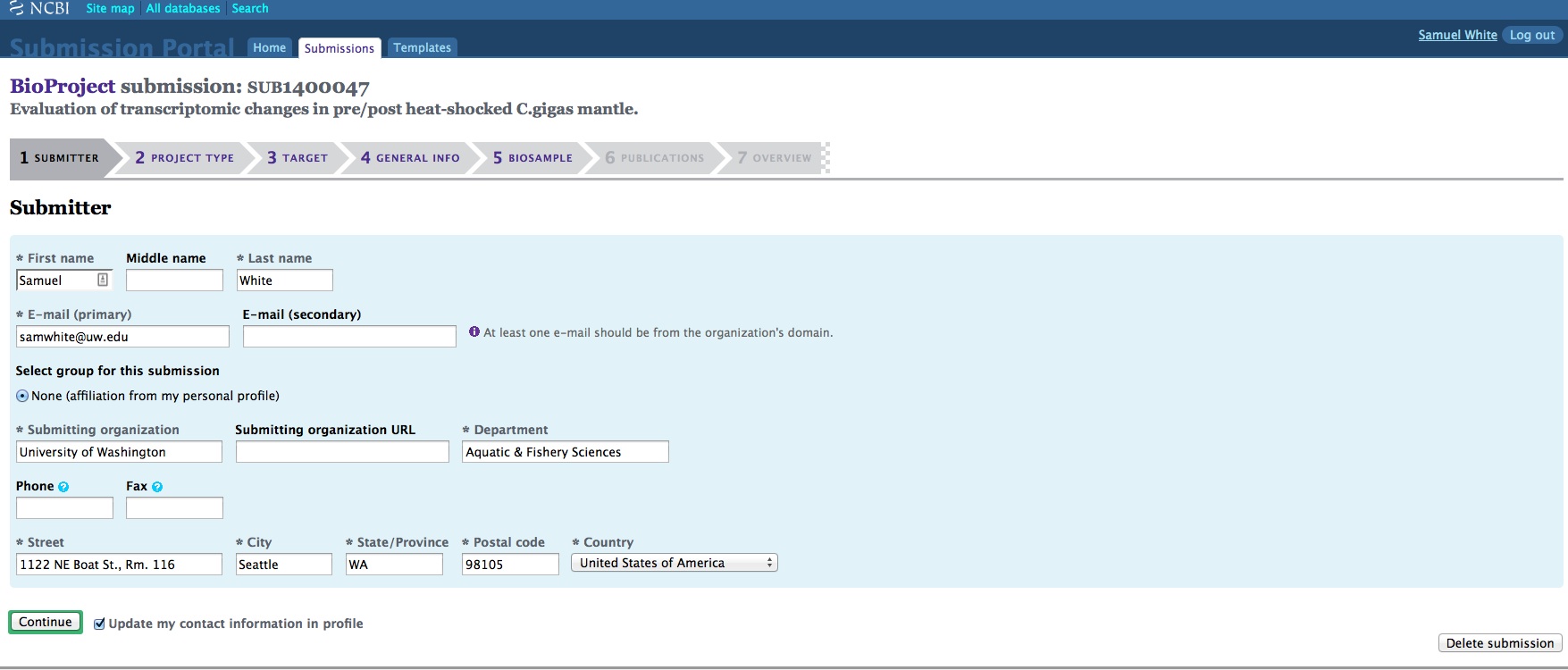
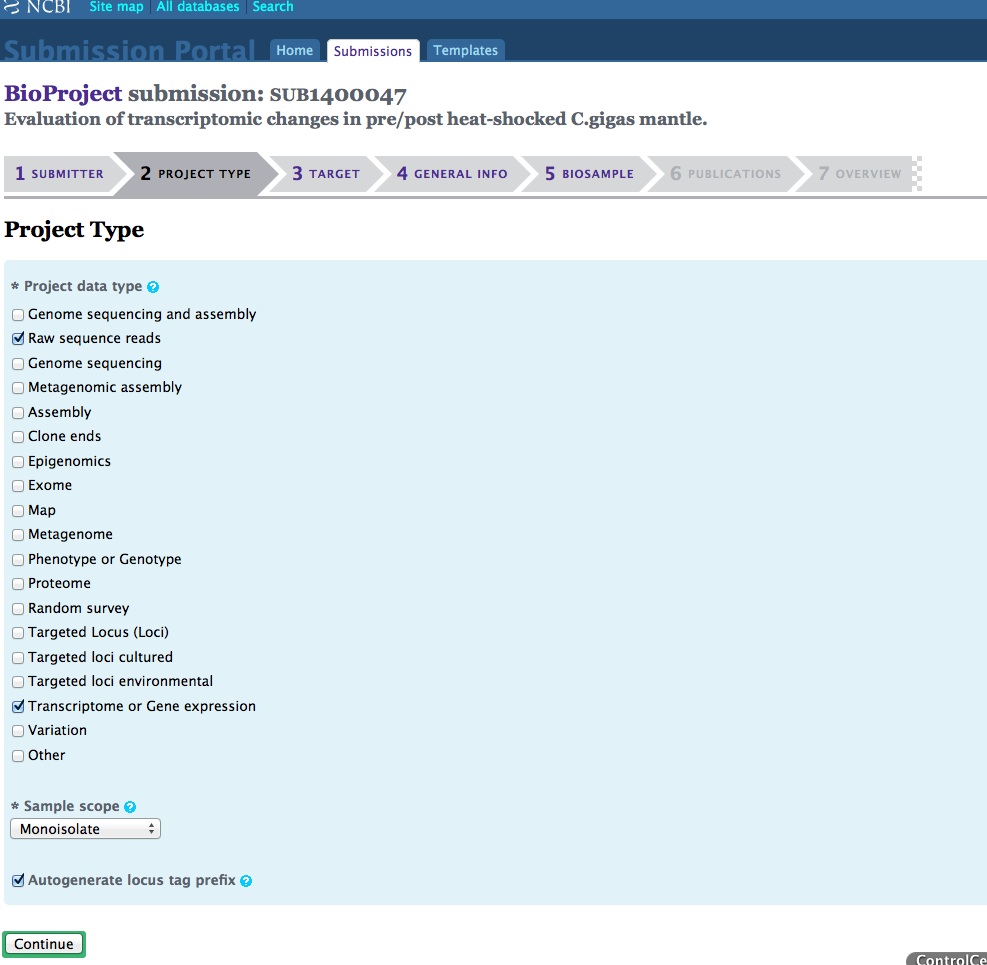
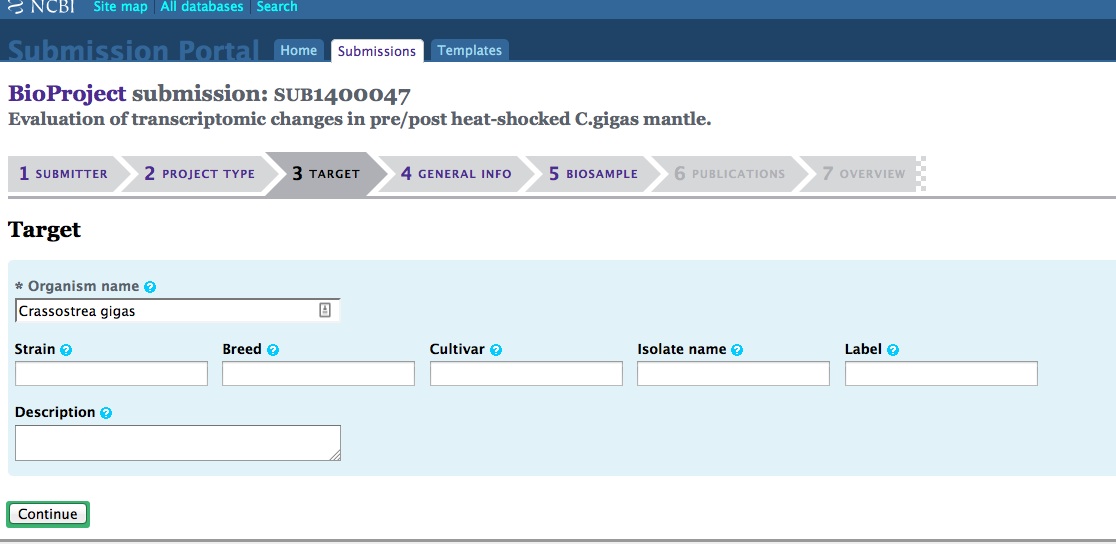
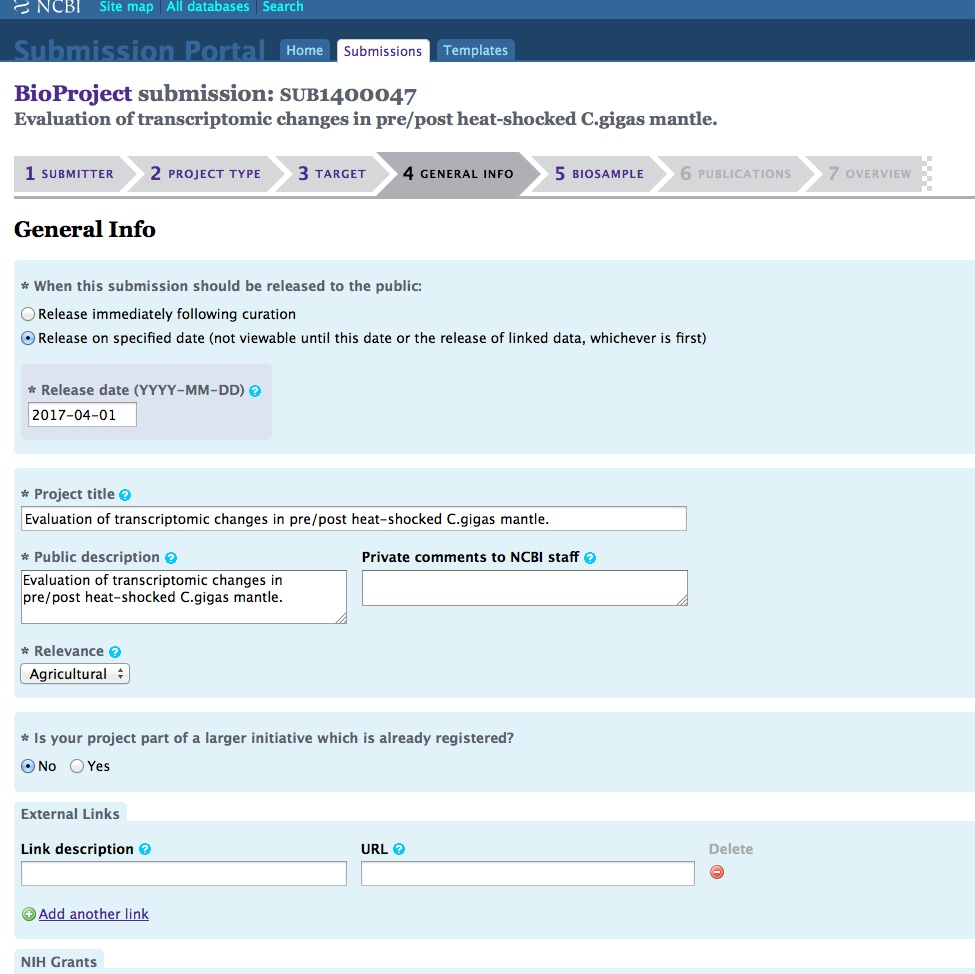
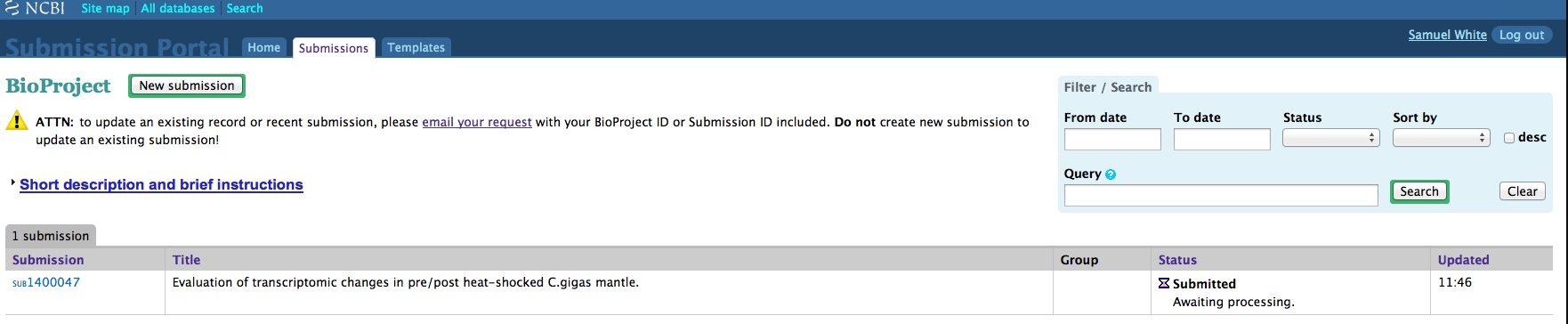
And, here are the screen caps, roughly in chronological order of how the process presents itself. It’s too time consuming to caption any of these, so I’m putting them up for a reference. Also, all of the information seen in these screen caps has been deleted (because the entire submission was totally jacked up in multiple facets), so don’t look for any of the various submission IDs - they no longer exist. This is really just to visually show how many steps there are in order to get stuff submitted - it’s brutal.