This isn’t really a notebook entry - it’s more of a traditional blog post.
It’s a quick summary of the frustrations and struggles I’ve encountered while trying to analyze the Olympia oyster GBS data. Hopefully it will serve as a place holder for others to find (and avoid) some of the pitfalls I’ve encountered so far. But, mostly, this is just for me to vent…
Using the Stacks program (on Hummingbird over the network to our server Owl) takes forever and, more importantly, consistently fails to complete the ustacks and cstacks programs.
Using the Stacks program (on Hummingbird via external HDD connected through Firewire) takes forever (combined, process_radtags and ustacks has been running since 20160428; that’s eight days)!!! Granted, this is running on all 96 samples, but, regardless, this type of time frame is not very conducive to productivity.
The “raw” non-demultiplexed fastq files supplied by BGI have a ‘N’ in the barcode in the FASTQ header lines. This prevents Stacks (and possibly Tassel - I’ll get to this in a second) from being able to perform the demultiplexing. Here’s a screen shot of what I’m talking about:
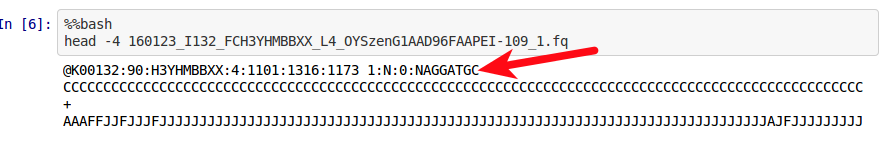 (http://owl.fish.washington.edu/Athaliana/20160503_BGI_GBS_fastq_header_barcode.png)
(http://owl.fish.washington.edu/Athaliana/20160503_BGI_GBS_fastq_header_barcode.png)
Cyverse has a program called Tassel that should be able to handle GBS data just like ours. However, it doesn’t produce the expected output to proceed to the second step. Although I haven’t tested it, it’s possible that the problem is related to the ‘N’ in the FASTQ header barcode sequence I mentioned above. I suspect it’s related because the first step in using Tassel is demultiplexing utilizing a supplied barcode keyfile.
Cyverse has Stacks installed, but in order to use it, someone has to build a Cyverse “app.” I’ve tried and the process is brutal. It’s not conducive for a program (that is really a suite of programs) like Stacks that has so many command line options and, depending on your input file types (e.g. “non-standard” Illumina filenames for paired-end sequencing), requires looping over filenames to specify corresponding file pairs.
Pyrad actually worked relatively well, but the VCF output file (for visualizing in the Integrative Genomics Viewer) has an ill-formed header that IGV won’t accept. Attempts at tweaking the header don’t seem to resolve the issue. Additionally, it’s not apparent in the output files if individuals get grouped, even though there is an option to specify which individuals should be grouped together.
And, the most frustrating thing of all???!!! I just realized how to handle the problematic barcodes in the FASTQ headers!! Instead of trying to alter the FASTQ files (which I’ve been messing around with over the past few days), all I’ve needed to do this entire time is CHANGE THE BARCODE KEY FILE THAT STACKS AND/OR TASSEL USES TO HAVE A ‘N’ AT THE BEGINNING OF EACH BARCODE!
I’m going to go cry now…
Regardless of that last one, it doesn’t change the fact that Stacks is painfully slow and, at times, unreliable.