We received whole genome bisulfite sequencing (WGBS) data from Genewiz last week on 20190408, so ran FastQC on the files on my computer (swoose). FastQC results will be added to Nightingales Google Sheet.
Each set of FastQs were processed with a bash script. This file (ends with .sh) can be found in each corresponding output folder (see below).
RESULTS
Output folders:
Roberto’s C.gigas
Yaamini’s C.gigas
Shelly’s P.generosa
Well, Shelly and Yaamini’s data look as expected for BSseq data.
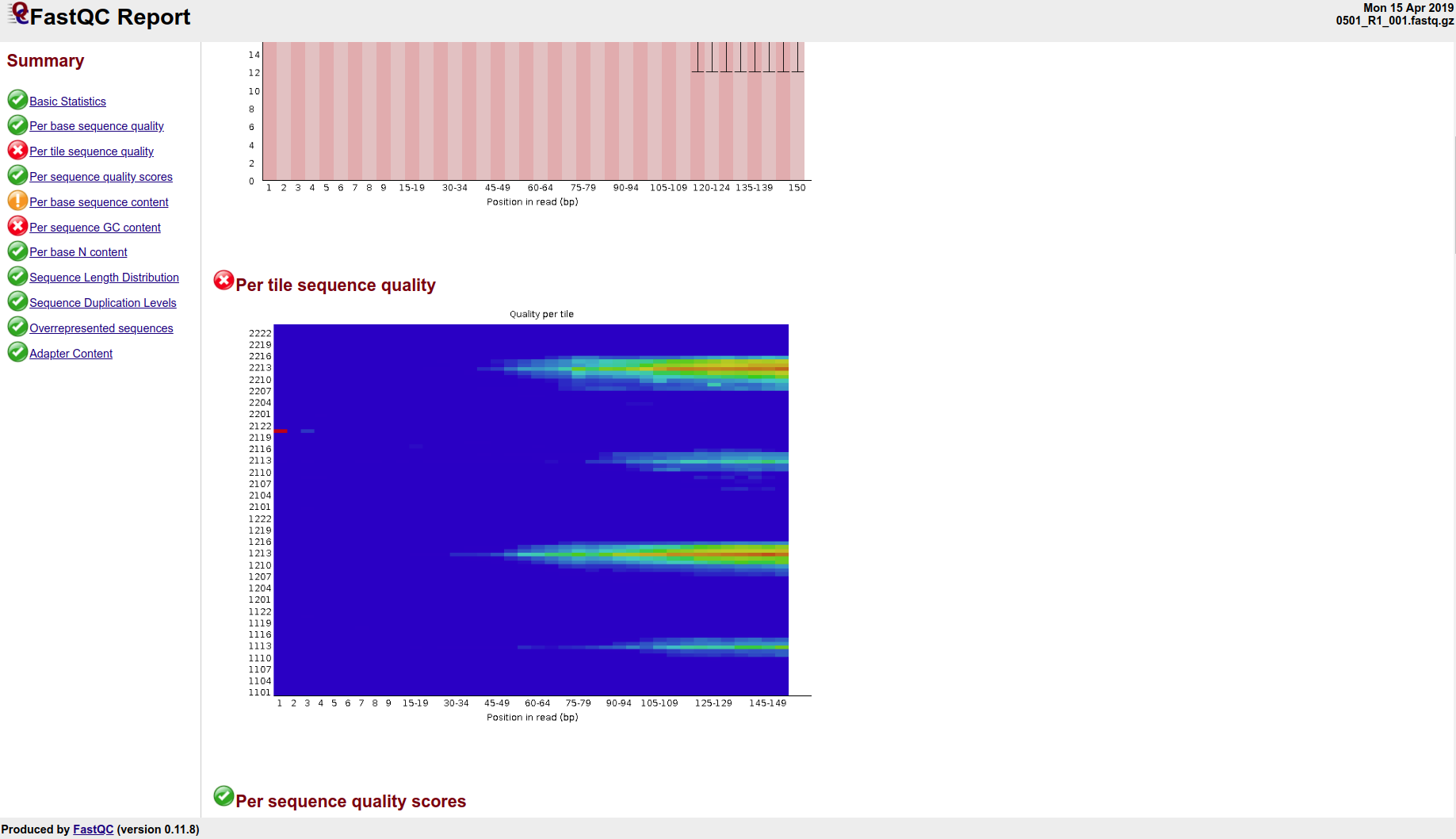
Roberto’s, however, does not look particularly good. All of his samples fail the “Per Tile Sequence Quality” test. I’m not sure I’ve ever seen sequences outright fail this before. Sure, we’ve had our share of sequences that might generate a warning, but not outright fail. And, it’s all of them! This suggests that something went wrong with the sequencer. This is idea is also partially supported by a message from Genewiz during the process that our data delivery date would be delayed due to a technical issue with the sequencer… However, I didn’t think they’d send us bad data. I’ve contacted them to see how to proceed.
I’ve also informed them that we sequenced with 30x coverage knowing that we’d lose a lot of data during the alignment process due the nature of the bisulfite-converted DNA and difficulties with aligning accurately. We did not anticipate having to discard a significant amount of sequencing reads due to poor quality. The combination of these two could bring our actual coverage below our desired minimum (5x).
Here’s a screenshot of one of Roberto’s samples (they all look like this, if not a bit worse):