Nearing the end of this quick metagenomics comparison of taxonomic differences between the two pH treatments (pH=7.1 and pH=8.2). Previously ran:
Here’s how the sample names breakdown:
| Sample | Develomental Stage (days post-fertilization) | pH Treatment |
|---|---|---|
| MG1 | 13 | 8.2 |
| MG2 | 17 | 8.2 |
| MG3 | 6 | 7.1 |
| MG5 | 10 | 8.2 |
| MG6 | 13 | 7.1 |
| MG7 | 17 | 7.1 |
After this completes, I’ll run KronaTools to get a rundown on taxonomic makeup of these two different pH treatments. I don’t expect BLASTn to take terribly long (based on previous metagenomics runs wit this data set); I’d guess around 6hrs.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=blastn_metagenomics
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=25-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/outputs/20190416_metagenomics_pgen_blastn
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
date >> system_path.log
echo "" >> system_path.log
echo "System PATH for $SLURM_JOB_ID" >> system_path.log
echo "" >> system_path.log
printf "%0.s-" {1..10} >> system_path.log
echo ${PATH} | tr : \\n >> system_path.log
wd="$(pwd)"
threads=28
# Paths to programs
blast_dir="/gscratch/srlab/programs/ncbi-blast-2.8.1+/bin"
blastn="${blast_dir}/blastn"
# Paths to blastdbs
blastdb_dir="/gscratch/srlab/blastdbs/ncbi-nr-nt-v5"
blast_db="${blastdb_dir}/nt"
# Directory with metagenemark FastAs
fasta_dir="/gscratch/scrubbed/samwhite/outputs/20190416_metagenomics_pgen_metagenemark"
# Export BLAST database directory
export BLASTDB=${blastdb_dir}
# Loop through metagenemark nucleotide FastAs
# Create list of those FastAs for reference
# Parse out sample names
# Run BLASTn on each FastA
for fasta in ${fasta_dir}/*nucleotides.fasta
do
echo ${fasta} >> input.fasta.list.txt
no_ext=${fasta%%.*}
sample_name=$(echo ${no_ext##*/})
# Run blastx on Trinity fasta
${blastn} \
-query ${fasta} \
-db ${blast_db} \
-max_target_seqs 1 \
-outfmt "6 std staxids" \
-evalue 1e-10 \
-num_threads ${threads} \
> ${wd}/${sample_name}.blastn.outfmt6
doneRESULTS
Runtime was ~9hrs for these.
Output folder:
pH=7.1 BLASTn Output:
547,999 matches
2,188,436 seqs in input FastA
~25% of input seqs were matched
pH=8.2 BLASTn Output:
298,564
2,047,907 seqs in input FastA
~15% of input seqs were matched
Interactive taxonomic Krona Plot (HTML):
Here are just a couple snapshots of the plots to add some “spice” to this notebook entry:
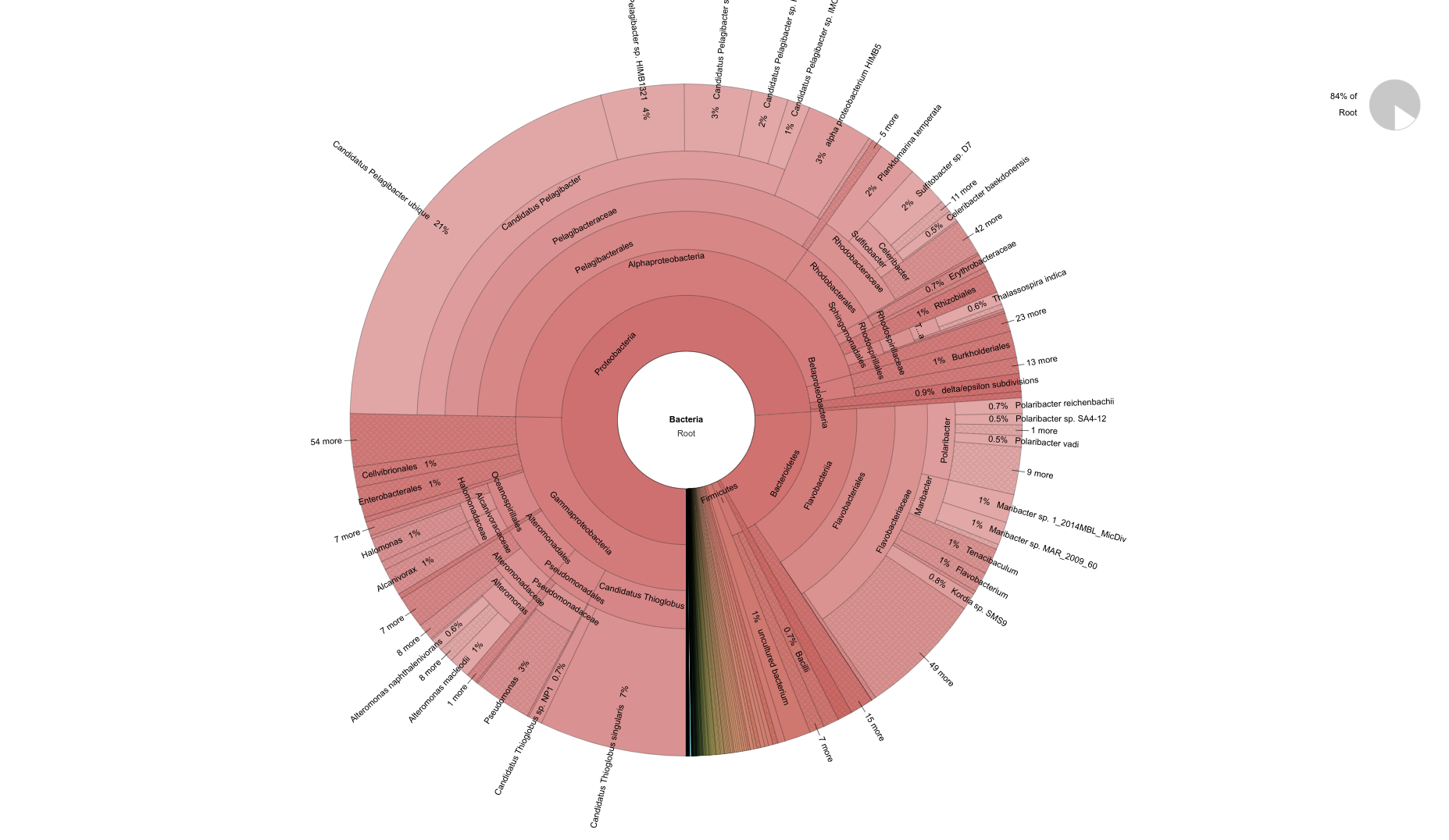
pH=7.1 Bacteria
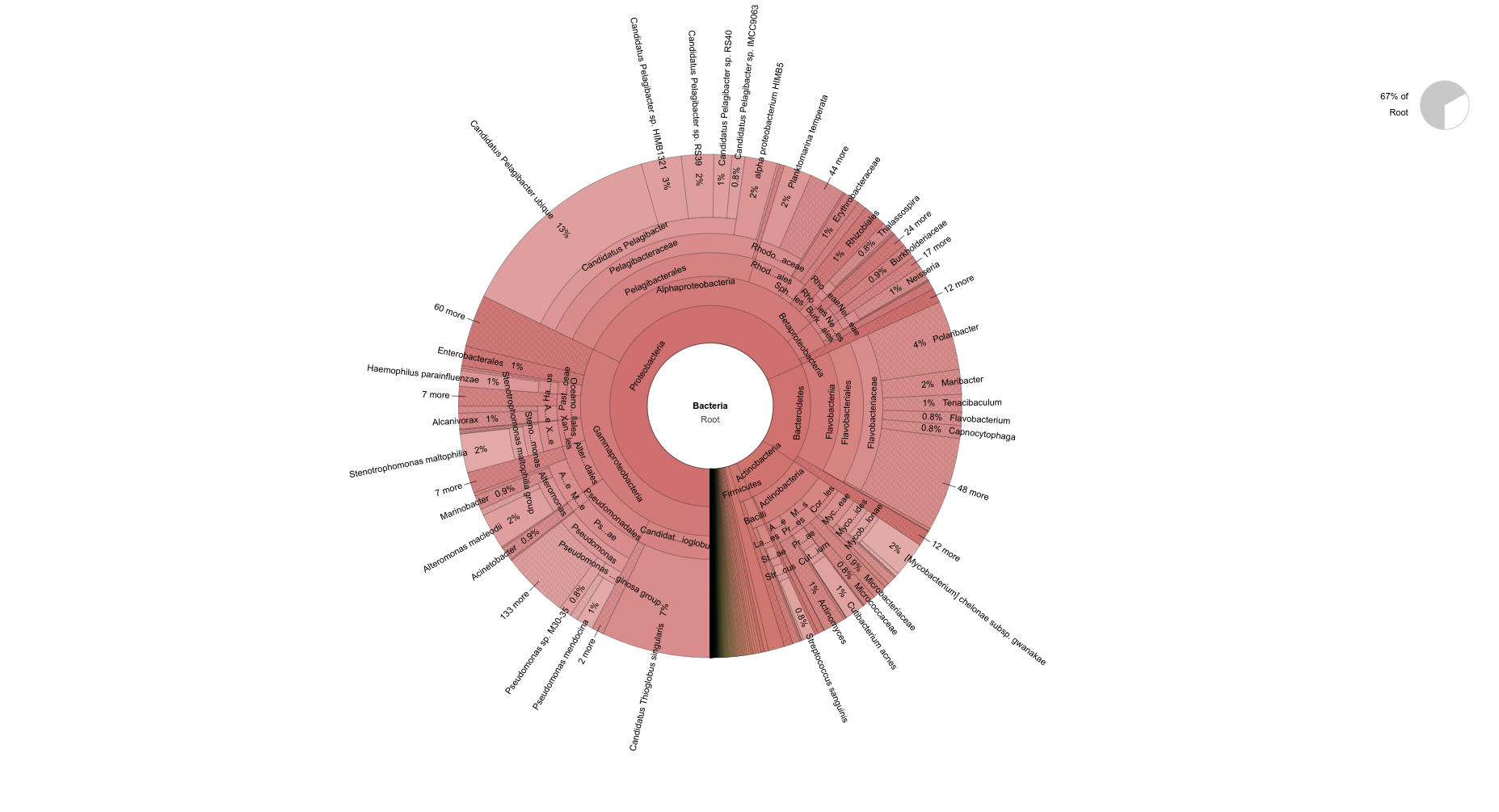
pH=8.2 Bacteria
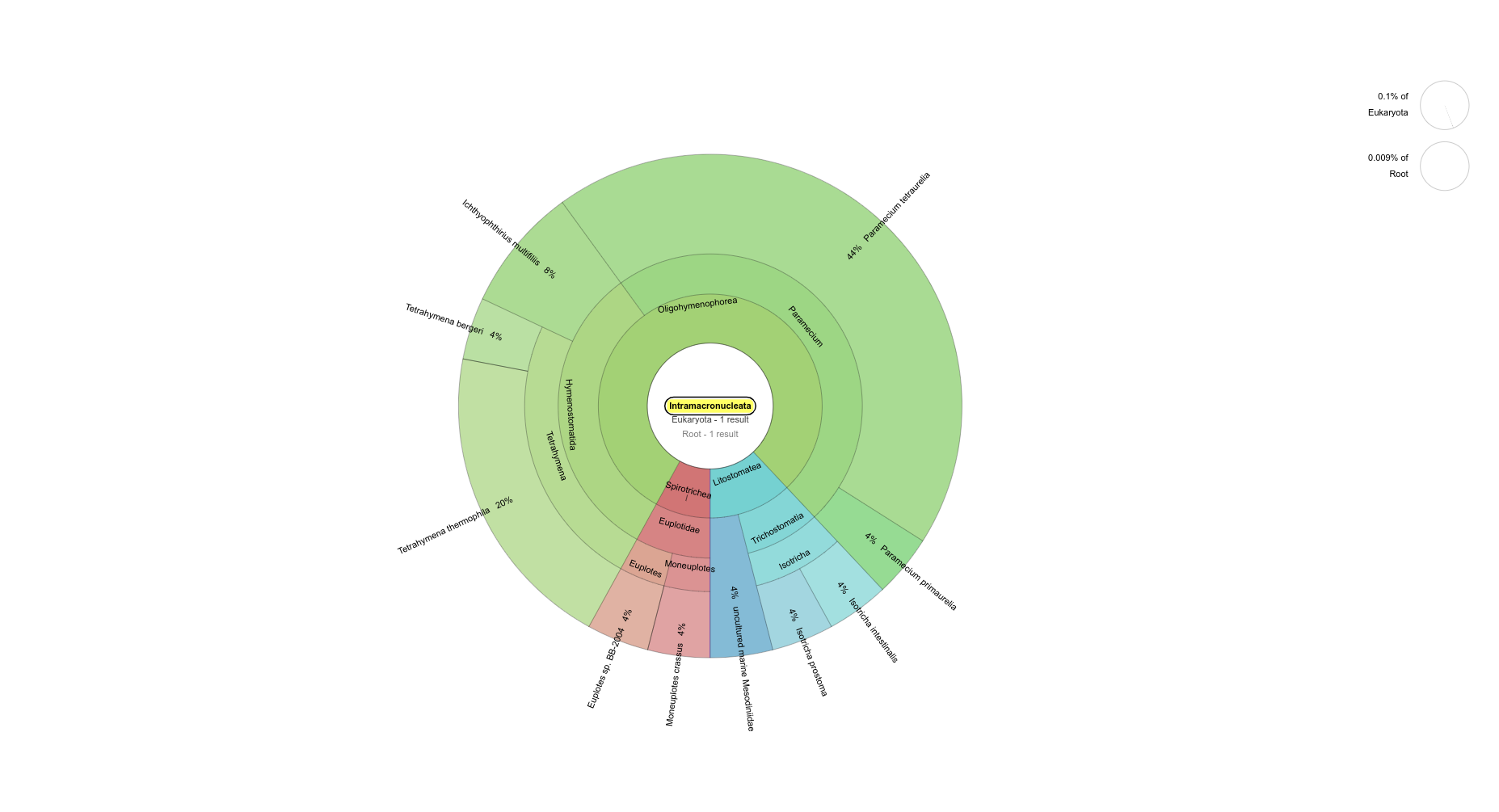
pH=7.1 Intramacronucleata (ciliate subphylum)
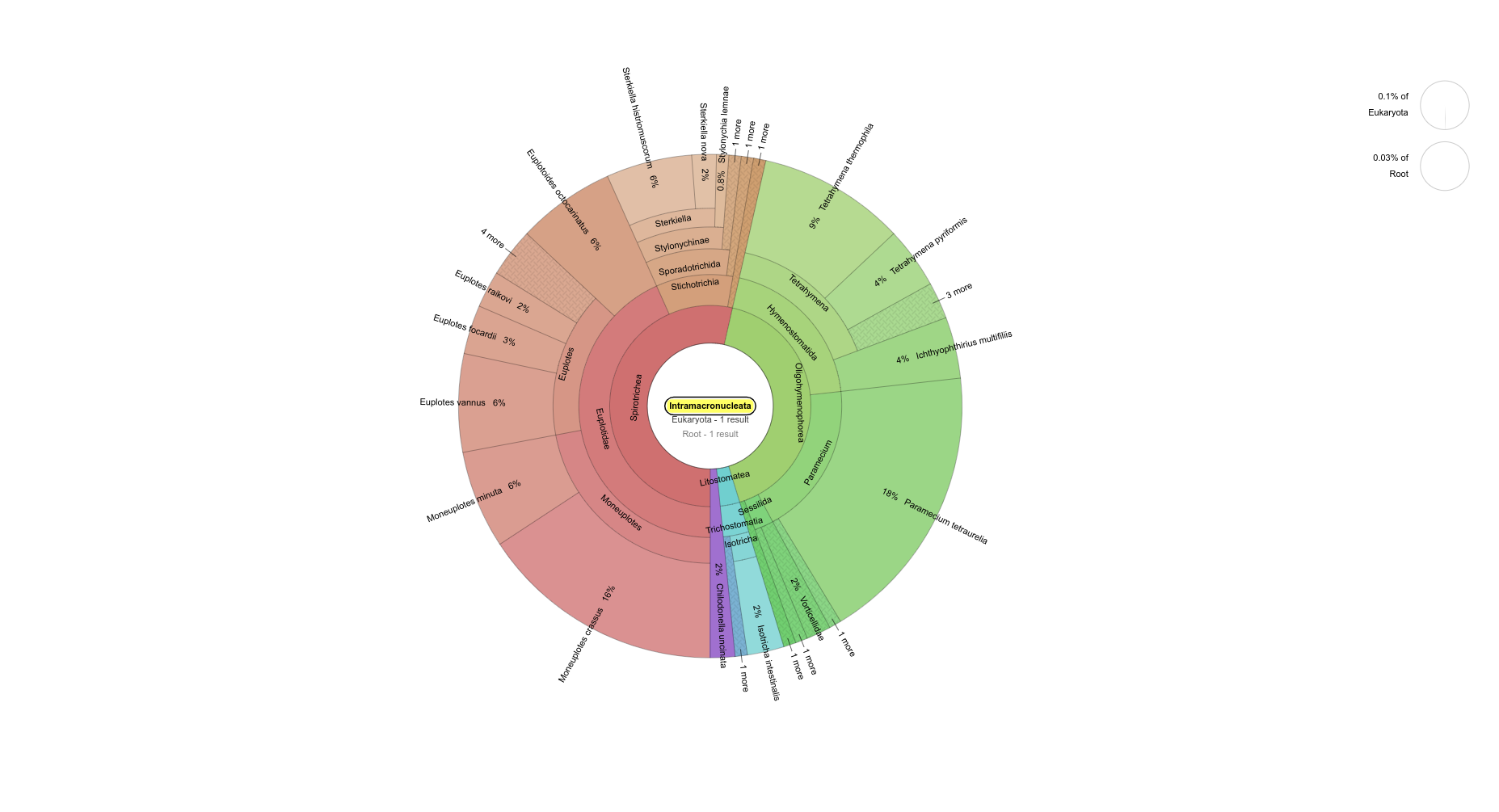
pH=8.2 Intramacronucleata (ciliate subphylum)