After a meeting on this project around the middle of May, we decided to try various approaches to assessing the metagenome. One aspect was to add coverage sequencing coverage information to our BLASTx taxonomy visualizations. I used the MEGAHIT coverage info from 20190327 and the subsequent BLASTx data from 20190516.
Briefly, I parsed out and joined the data to generate the appropriate input file needed for visualizations using Krona Tools and then ran the ktImportTaxonomy Krona Tools program. This is all detailed in the Jupyter Notebook below.
Here’s how the sample names breakdown:
| Sample | Develomental Stage (days post-fertilization) | pH Treatment |
|---|---|---|
| MG1 | 13 | 8.2 |
| MG2 | 17 | 8.2 |
| MG3 | 6 | 7.1 |
| MG5 | 10 | 8.2 |
| MG6 | 13 | 7.1 |
| MG7 | 17 | 7.1 |
Jupyter Notebook (GitHub):
NBViewer for viewing notebook:
RESULTS
Output folder:
Krona Plots (HTML):
The Krona plot is interactive and allows the user to select the different plots that they want to see and then “drill down” further in to the various taxonomies. Unfortunately, the “Avg. score” (i.e. average of the average sequencing coverage) is only displayed as a number in the upper right corner. There’s no color coding. Well, this isn’t entirely true. An option is availabe to “Color by Avg. Score”, however it seems that the color range is a default and does not dynamically adjust to the input ranges. As such, in this particular case, the coverage is all fairly low in the range (~20 fold coverage) and simply all gets colored the same, since the default score range is 0.3 - 6367.
Example of Krona plot showing the Avg. score (i.e. the average of average sequencing coverage for a given taxonomic group):
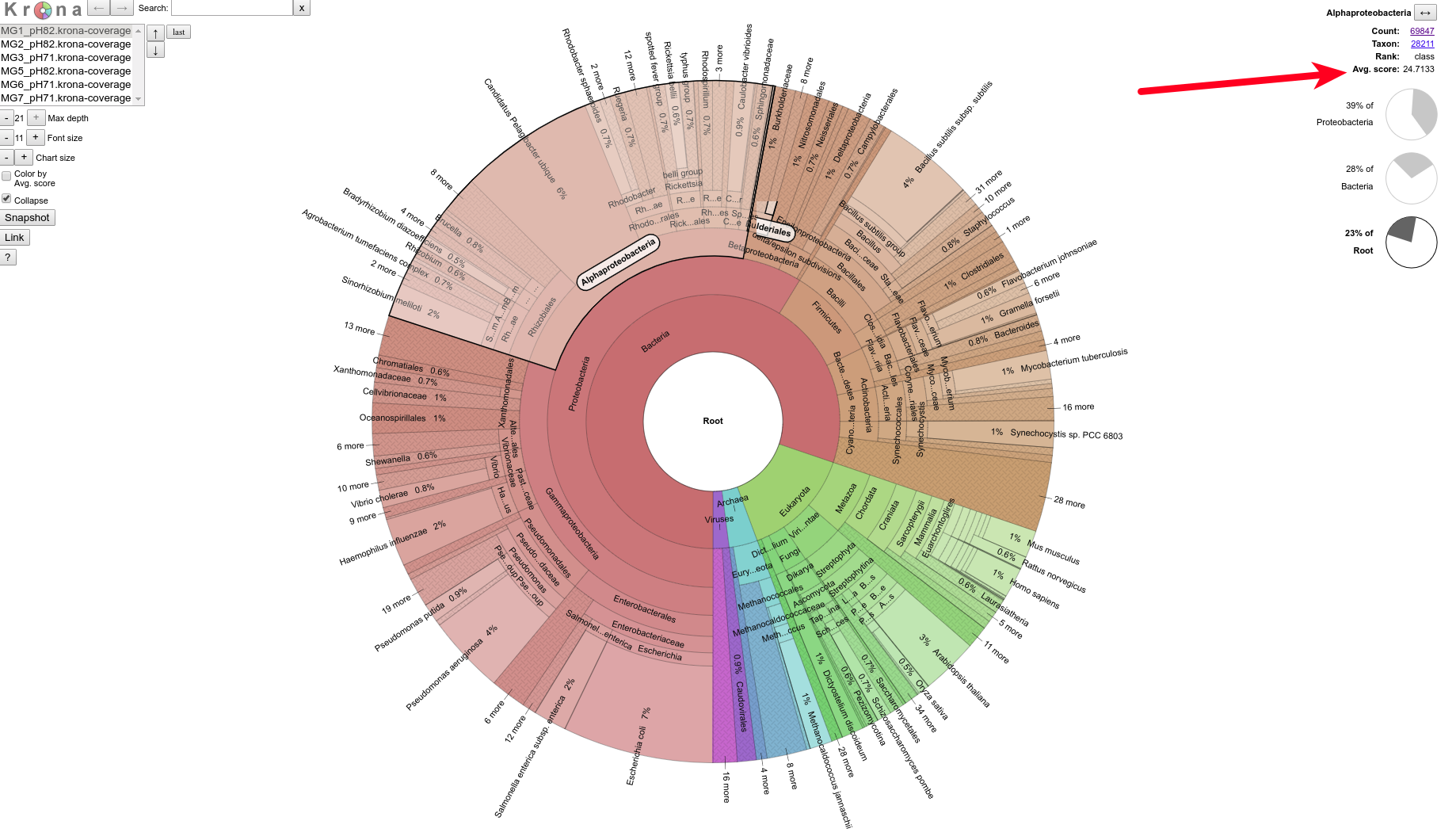
Although I still haven’t figured out a way to actually pull out any of the data being used to generate the plots, a quick clicking around revealed a couple of things:
Average coverage across taxa within a given data set appears to be relatively equal.
Average coverage across taxa across most data sets appears to be ~20x.
MG3 (pH = 7.1) sample exhibits ~50% less average coverage across taxa: ~10X
MG3 (pH = 7.1) sample shows higher average coverage in Eukaryotes than Bacteria, yet all other samples are the opposite.
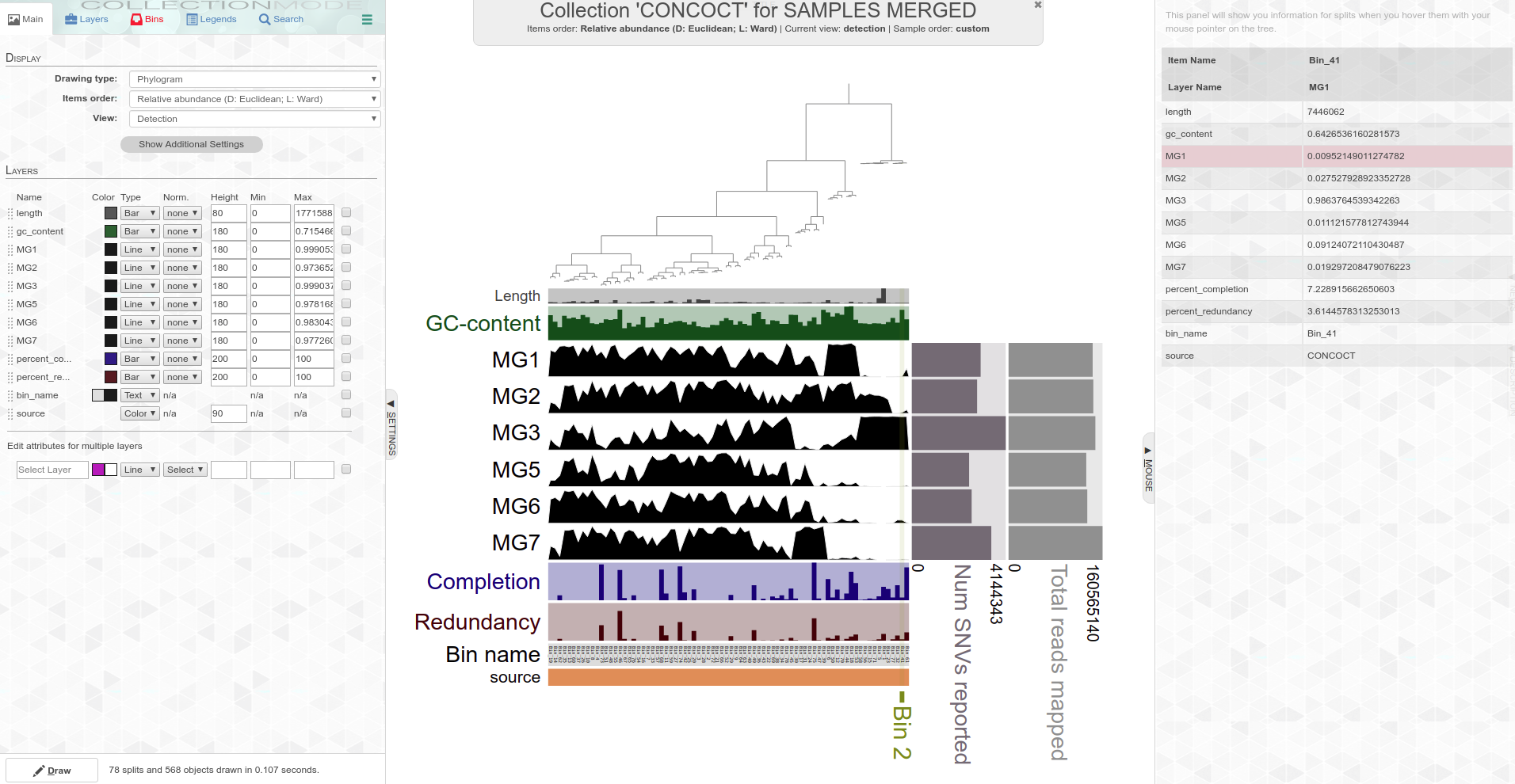
It seems that MG3 (pH = 7.1) is a bit of an anomaly. Analysis I did with Anvi’o (using CONCOCT for genome abundance determination) on 20190401, also shows that MG3 is noticeably different than the other five samples:
We have a meeting tomorrow (20190613) to go over this project with Emma. Should be good to determine what direction we will take and produce further ideas for analysis.