Earlier today, I generated the necessary Hista2 index, which incorporated splice sites and exons, for use with Stringtie in order to identify transcript isoforms in our Olurida_v081 annotation.
I used all the trimmed FastQ files from the 20180827 Trinity transcriptome assembly, as this utilized all of our existing RNAseq data.
Command to pull trimmed files (Trimmomatic) out of the Trinity output folder that is a gzipped tarball:
tar -ztvf trinity_out_dir.tar.gz \
| grep -E "*P.qtrim.gz" \
&& tar -zxvf trinity_out_dir.tar.gz \
-C /home/sam/Downloads/ \
--wildcards "*P.qtrim.gz"This was run locally on my computer (swoose) and then rsync’d to Mox.
NOTE: The “P” in the *.P.qtrim.gz represents trimmed reads that are properly paired, as determined by Trimmomatic/Trinity. See the fastq.list.txt for the list of FastQ files used as input. For more info on input FastQ files, refer to the Nightingales Google Sheet.
Here’s the quick rundown of how transcript isoform annotation with Stringtie runs:
Use Hisat2 reference index with identified splice sites and exons (this was done yesterday).
Use Hisat2 to create alignments from each pair of trimmed FastQ files. Need to use the
--downstream-transcriptome-assemblyoption!!!Use Stringtie to create a GTF for each alignment.
Use Stringtie to create a singular, merged GTF from all of the individual GTFs.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=oly_stringtie
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=25-00:00:00
## Memory per node
#SBATCH --mem=500G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/outputs/20190625_stringtie_oly_v081
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
date >> system_path.log
echo "" >> system_path.log
echo "System PATH for $SLURM_JOB_ID" >> system_path.log
echo "" >> system_path.log
printf "%0.s-" {1..10} >> system_path.log
echo "${PATH}" | tr : \\n >> system_path.log
threads=27
genome_index_name="Olurida_v081"
# Paths to programs
hisat2_dir="/gscratch/srlab/programs/hisat2-2.1.0"
hisat2="${hisat2_dir}/hisat2"
samtools="/gscratch/srlab/programs/samtools-1.9/samtools"
stringtie="/gscratch/srlab/programs/stringtie-1.3.6.Linux_x86_64/stringtie"
# Input/output files
genome_gff="/gscratch/srlab/sam/data/O_lurida/genomes/Olurida_v081/20181127_oly_genome_snap02.all.renamed.putative_function.domain_added.gff"
genome_index_dir="/gscratch/srlab/sam/data/O_lurida/genomes/Olurida_v081"
fastq_dir="/gscratch/srlab/sam/data/O_lurida/RNAseq/"
gtf_list="gtf_list.txt"
## Inititalize arrays
fastq_array_R1=()
fastq_array_R2=()
names_array=()
# Copy Hisat2 genome index
rsync -av "${genome_index_dir}"/Olurida_v081*.ht2 .
# Create array of fastq R1 files
for fastq in ${fastq_dir}*R1*.gz
do
fastq_array_R1+=(${fastq})
done
# Create array of fastq R2 files
for fastq in ${fastq_dir}*R2*.gz
do
fastq_array_R2+=(${fastq})
done
# Create array of sample names
## Uses parameter substitution to strip leading path from filename
## Uses awk to parse out sample name from filename
for R1_fastq in ${fastq_dir}*R1*.gz
do
names_array+=($(echo ${R1_fastq#${fastq_dir}} | awk -F"[_.]" '{print $1 "_" $5}'))
done
# Create list of fastq files used in analysis
## Uses parameter substitution to strip leading path from filename
for fastq in ${fastq_dir}*.gz
do
echo "${fastq#${fastq_dir}}" >> fastq.list.txt
done
# Hisat2 alignments
for index in "${!fastq_array_R1[@]}"
do
sample_name=$(echo "${names_array[index]}")
"${hisat2}" \
-x "${genome_index_name}" \
--downstream-transcriptome-assembly \
-1 "${fastq_array_R1[index]}" \
-2 "${fastq_array_R2[index]}" \
-S "${sample_name}".sam \
2> "${sample_name}"_hisat2.err
# Sort SAM files, convert to BAM, and index
"${samtools}" view \
-@ "${threads}" \
-Su "${sample_name}".sam \
| "${samtools}" sort - \
-@ "${threads}" \
-o "${sample_name}".sorted.bam
"${samtools}" index "${sample_name}".sorted.bam
# Run stringtie on alignments
"${stringtie}" "${sample_name}".sorted.bam \
-p "${threads}" \
-o "${sample_name}".gtf \
-G "${genome_gff}" \
-C "${sample_name}.cov_refs.gtf"
# Add GTFs to list file
echo "${sample_name}.gtf" >> "${gtf_list}"
done
# Create singular transcript file, using GTF list file
"${stringtie}" --merge \
"${gtf_list}" \
-p "${threads}" \
-G "${genome_gff}" \
-o "${genome_index_name}".stringtie.gtf
# Delete unneccessary index files
rm Olurida_v081*.ht2
# Delete unneded SAM files
rm *.samRESULTS
This took about one day to run:

Output folder:
Merged GTF:
Although I won’t link them here, each input FastQ pair has a corresponding alignment file (BAM), coverage file (.cov_refs.gtf), Hisat2 alignment stats file (_hisat2.err), and GTF.
Below, I’ve included a screencap of an Integrated Genomics Viewer (IGV) session of a contig that shows mRNAs identified via the MAKER annotation, along with the Stringtie GTF:
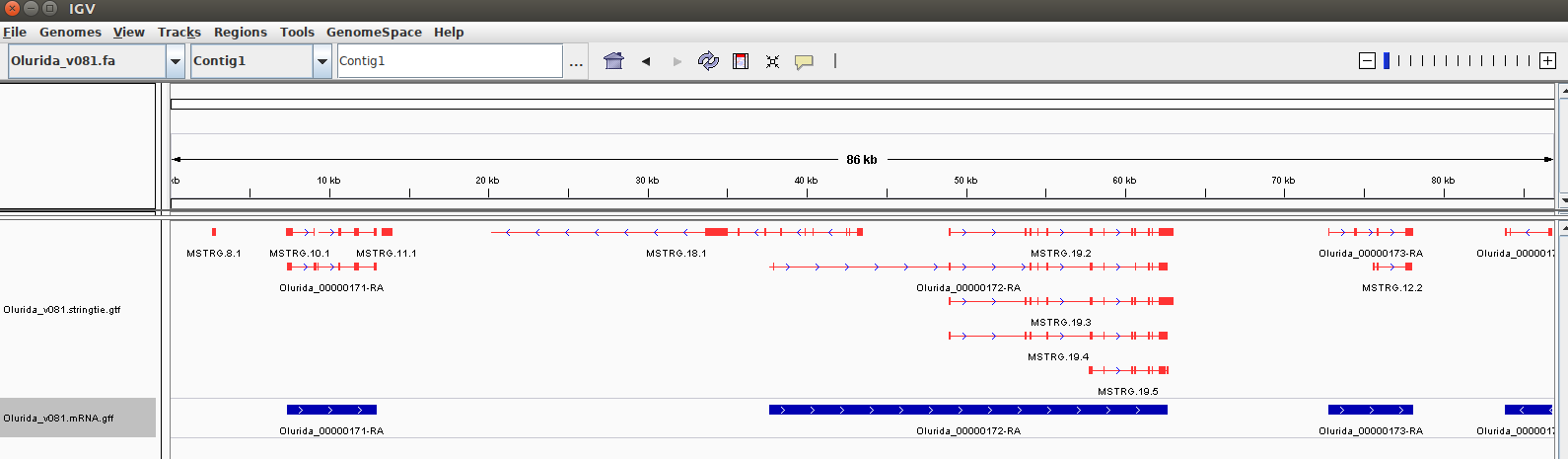
The blue tracks correspond to the mRNAs identified by MAKER. The red tracks correspond to transcripts identified by Stringtie. The horizontal lines in the GTF track represent the length of an identified transcript. Each vertical line/block in the GTF track represents an exon. This provides an excellent visualization of potential isoforms identified by Stringtie within a given mRNA!
A couple of things to also point out in this example:
There is an mRNA that appears to have no isoforms (Olurida_00000174-RA; on far right of image with name truncated). It’s always good to see something like this to make you believe the data, as not all mRNAs should have isoforms.
A “rogue” transcript (MSTRG.8.1; on the far left of the image). This doesn’t line up with an mRNA feature from the MAKER annotation, nor does it have any associated directionality. Is this a mis-identified exon by Stringtie or a missed annotation by MAKER? Or, is it a partial annotation by Stringtie and MAKER didn’t mark it because of insufficient sequence for this end of this contig?
A transcript that is on the opposite strand (MSTRG.18.1). This doesn’t have a corresponding mRNA segment from the MAKER annotation. Is this “real”?
Anyway, just some things to think about. Will add results to the Slack thread that prompted this whole thing.
Would be good to visualize with other annotation tracks (e.g. CDS, TEs, etc) to see how things match up.