I previously created a subset of the Pgenerosa_v070 genome assembly that contains just the largest 18 scaffolds (these scaffolds were produced by Phase Genomics, utilizing some Hi-C sequencing). The new subsetted genome is labeled as Pgenerosa_v074.fa (914MB).
As part of that, Steven wanted this version annotated using MAKER.
This will perform the following:
- one round of MAKER gene model predictions
- two rounds of SNAP gene model training/predictions
- renaming of gene models to NCBI-standardized convention
- functional characterization of protein models (via BLASTp)
- functional characterization of protein domains (via InterProScan5)
Here are a list of the input files used for the various components of the MAKER annotation:
Transcriptome FastA files (links to notebook entries):
NCBI Protein FastA files
NCBI Crassostrea gigas proteome (downloaded 20181119):
GCA_000297895.1_oyster_v9_protein.faaNCBI Crassostrea virginica proteome (downloaded 20181119):
GCF_002022765.2_C_virginica-3.0_protein.faaSwissProt BLASTp database(downloaded 20190109): uniprot_sprot.fasta
TransDecoder protein FastA files (links to notebook entries)
Repeats Files
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=maker_pgen074
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=2
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=40-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --workdir=/gscratch/scrubbed/samwhite/outputs/20190701_pgen_maker_v074_annotation
# Exit if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Load Open MPI module for parallel, multi-node processing
module load icc_19-ompi_3.1.2
# SegFault fix?
export THREADS_DAEMON_MODEL=1
# Document programs in PATH (primarily for program version ID)
date >> system_path.log
echo "" >> system_path.log
echo "System PATH for $SLURM_JOB_ID" >> system_path.log
echo "" >> system_path.log
printf "%0.s-" {1..10} >> system_path.log
echo "${PATH}" | tr : \\n >> system_path.log
# Add BLAST to system PATH
export PATH=$PATH:/gscratch/srlab/programs/ncbi-blast-2.6.0+/bin
export BLASTDB=/gscratch/srlab/blastdbs/UniProtKB_20181008
## Establish variables for more readable code
wd=$(pwd)
maker_dir=/gscratch/srlab/programs/maker-2.31.10/bin
snap_dir=/gscratch/srlab/programs/maker-2.31.10/exe/snap
### Paths to Maker binaries
maker=${maker_dir}/maker
gff3_merge=${maker_dir}/gff3_merge
maker2zff=${maker_dir}/maker2zff
fathom=${snap_dir}/fathom
forge=${snap_dir}/forge
hmmassembler=${snap_dir}/hmm-assembler.pl
fasta_merge=${maker_dir}/fasta_merge
map_ids=${maker_dir}/maker_map_ids
map_gff_ids=${maker_dir}/map_gff_ids
map_fasta_ids=${maker_dir}/map_fasta_ids
functional_fasta=${maker_dir}/maker_functional_fasta
functional_gff=${maker_dir}/maker_functional_gff
ipr_update_gff=${maker_dir}/ipr_update_gff
iprscan2gff3=${maker_dir}/iprscan2gff3
blastp_dir=${wd}/blastp_annotation
maker_blastp=${wd}/blastp_annotation/blastp.outfmt6
maker_prot_fasta=${wd}/snap02/Pgenerosa_v074_snap02.all.maker.proteins.fasta
maker_prot_fasta_renamed=${wd}/snap02/Pgenerosa_v074_snap02.all.maker.proteins.renamed.fasta
maker_transcripts_fasta=${wd}/snap02/Pgenerosa_v074_snap02.all.maker.transcripts.fasta
maker_transcripts_fasta_renamed=${wd}/snap02/Pgenerosa_v074_snap02.all.maker.transcripts.renamed.fasta
snap02_gff=${wd}/snap02/Pgenerosa_v074_snap02.all.gff
snap02_gff_renamed=${wd}/snap02/Pgenerosa_v074_snap02.all.renamed.gff
put_func_gff=Pgenerosa_v074_genome_snap02.all.renamed.putative_function.gff
put_func_prot=Pgenerosa_v074_genome_snap02.all.maker.proteins.renamed.putative_function.fasta
put_func_trans=Pgenerosa_v074_genome_snap02.all.maker.transcripts.renamed.putative_function.fasta
put_domain_gff=Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff
ips_dir=${wd}/interproscan_annotation
ips_base=Pgenerosa_v074_maker_proteins_ips
ips_name=Pgenerosa_v074_maker_proteins_ips.tsv
id_map=${wd}/snap02/Pgenerosa_v074_genome.map
ips_domains=Pgenerosa_v074_genome_snap02.all.renamed.visible_ips_domains.gff
## Path to blastp
blastp=/gscratch/srlab/programs/ncbi-blast-2.6.0+/bin/blastp
## Path to InterProScan5
interproscan=/gscratch/srlab/programs/interproscan-5.31-70.0/interproscan.sh
## Store path to options control file
maker_opts_file=./maker_opts.ctl
### Path to genome FastA file
genome=/gscratch/srlab/sam/data/P_generosa/genomes/Pgenerosa_v074.fa
### Paths to transcriptome FastA files
ctendia_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/ctenidia/Trinity.fasta
gonad_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/gonad/Trinity.fasta
heart_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/heart/Trinity.fasta
EPI99_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/larvae/EPI99/Trinity.fasta
EPI115_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/juvenile/EPI115/Trinity.fasta
EPI116_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/juvenile/EPI116/Trinity.fasta
EPI123_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/juvenile/EPI123/Trinity.fasta
EPI124_transcriptome=/gscratch/srlab/sam/data/P_generosa/transcriptomes/juvenile/EPI124/Trinity.fasta
### Path to Crassotrea gigas NCBI protein FastA
gigas_proteome=/gscratch/srlab/sam/data/C_gigas/gigas_ncbi_protein/GCA_000297895.1_oyster_v9_protein.faa
### Path to Crassostrea virginica NCBI protein FastA
virginica_proteome=/gscratch/srlab/sam/data/C_virginica/virginica_ncbi_protein/GCF_002022765.2_C_virginica-3.0_protein.faa
### Path to Panopea generosa TransDecoder protein FastAs
panopea_td_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/20180827_trinity_geoduck.fasta.transdecoder.pep
pgen_td_ctenidia_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/ctenidia/Trinity.fasta.transdecoder.pep
pgen_td_larvae_EPI99_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/larvae/EPI99/Trinity.fasta.transdecoder.pep
pgen_td_juv_EPI115_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/juvenile/EPI115/Trinity.fasta.transdecoder.pep
pgen_td_juv_EPI116_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/juvenile/EPI116/Trinity.fasta.transdecoder.pep
pgen_td_juv_EPI123_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/juvenile/EPI123/Trinity.fasta.transdecoder.pep
pgen_td_juv_EPI124_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/juvenile/EPI124/Trinity.fasta.transdecoder.pep
pgen_td_gonad_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/gonad/Trinity.fasta.transdecoder.pep
pgen_td_heart_proteome=/gscratch/srlab/sam/data/P_generosa/proteomes/heart/Trinity.fasta.transdecoder.pep
### Path to P.generosa-specific RepeatModeler library
repeat_library=/gscratch/srlab/sam/data/P_generosa/repeats/Pgenerosa_v074-families.fa
### Path to P.generosa-specific RepeatMasker GFF
rm_gff=/gscratch/srlab/sam/data/P_generosa/repeats/Pgenerosa_v074.fa.out.gff
### Path to SwissProt database for BLASTp
sp_db_blastp=/gscratch/srlab/blastdbs/UniProtKB_20190109/uniprot_sprot.fasta
## Make directories
mkdir blastp_annotation
mkdir interproscan_annotation
mkdir snap01
mkdir snap02
## Create Maker control files needed for running Maker, only if it doesn't already exist and then edit it.
### Edit options file
### Set paths to P.generosa genome and transcriptome.
### Set path to combined C. gigas, C.virginica, P.generosa proteomes.
### The use of the % symbol sets the delimiter sed uses for arguments.
### Normally, the delimiter that most examples use is a slash "/".
### But, we need to expand the variables into a full path with slashes, which screws up sed.
### Thus, the use of % symbol instead (it could be any character that is NOT present in the expanded variable; doesn't have to be "%").
if [ ! -e maker_opts.ctl ]; then
$maker -CTL
sed -i "/^genome=/ s% %$genome %" "$maker_opts_file"
# Set transcriptomes to use
sed -i "/^est=/ s% %\
${ctendia_transcriptome},\
${gonad_transcriptome},\
${heart_transcriptome},\
${EPI99_transcriptome},\
${EPI115_transcriptome},\
${EPI116_transcriptome},\
${EPI123_transcriptome},\
${EPI124_transcriptome} %" \
"$maker_opts_file"
# Set proteomes to use
sed -i "/^protein=/ s% %\
${gigas_proteome},\
${panopea_td_proteome},\
${pgen_td_ctenidia_proteome},\
${pgen_td_gonad_proteome},\
${pgen_td_heart_proteome},\
${pgen_td_juv_EPI115_proteome},\
${pgen_td_juv_EPI116_proteome},\
${pgen_td_juv_EPI123_proteome},\
${pgen_td_juv_EPI124_proteome},\
${pgen_td_larvae_EPI99_proteome},\
${virginica_proteome} \
%" \
"$maker_opts_file"
# Set RepeatModeler library to use
sed -i "/^rmlib=/ s% %$repeat_library %" "$maker_opts_file"
# Set RepeatMasker GFF to use
sed -i "/^rm_gff=/ s% %${rm_gff} %" "$maker_opts_file"
# Set est2ggenome to 1 - tells MAKER to use transcriptome FastAs
sed -i "/^est2genome=0/ s/est2genome=0/est2genome=1/" "$maker_opts_file"
# Set protein2genome to 1 - tells MAKER to use protein FastAs
sed -i "/^protein2genome=0/ s/protein2genome=0/protein2genome=1/" "$maker_opts_file"
fi
## Run Maker
### Specify number of nodes to use.
mpiexec -n 56 $maker
## Merge gffs
${gff3_merge} -d Pgenerosa_v074.maker.output/Pgenerosa_v074_master_datastore_index.log
## GFF with no FastA in footer
${gff3_merge} -n -s -d Pgenerosa_v074.maker.output/Pgenerosa_v074_master_datastore_index.log > Pgenerosa_v074.maker.all.noseqs.gff
## Merge all FastAs
${fasta_merge} -d Pgenerosa_v074.maker.output/Pgenerosa_v074_master_datastore_index.log
## Extract GFF alignments for use in subsequent MAKER rounds
### Transcript alignments
awk '{ if ($2 == "est2genome") print $0 }' Pgenerosa_v074.maker.all.noseqs.gff > Pgenerosa_v074.maker.all.noseqs.est2genome.gff
### Protein alignments
awk '{ if ($2 == "protein2genome") print $0 }' Pgenerosa_v074.maker.all.noseqs.gff > Pgenerosa_v074.maker.all.noseqs.protein2genome.gff
### Repeat alignments
awk '{ if ($2 ~ "repeat") print $0 }' Pgenerosa_v074.maker.all.noseqs.gff > Pgenerosa_v074.maker.all.noseqs.repeats.gff
## Run SNAP training, round 1
cd "${wd}"
cd snap01
${maker2zff} ../Pgenerosa_v074.all.gff
${fathom} -categorize 1000 genome.ann genome.dna
${fathom} -export 1000 -plus uni.ann uni.dna
${forge} export.ann export.dna
${hmmassembler} genome . > Pgenerosa_v074_snap01.hmm
## Initiate second Maker run.
### Copy initial maker control files and
### Default gene prediction settings are 0 (i.e. don't generate Maker gene predictions)
### - use GFF subsets generated in first round of MAKER
### - set location of snaphmm file to use for gene prediction
### Percent symbols used below are the sed delimiters, instead of the default "/",
### due to the need to use file paths.
if [ ! -e maker_opts.ctl ]; then
$maker -CTL
sed -i "/^genome=/ s% %$genome %" maker_opts.ctl
# Set transcriptomes to use
sed -i "/^est=/ s% %\
${ctendia_transcriptome},\
${gonad_transcriptome},\
${heart_transcriptome},\
${EPI99_transcriptome},\
${EPI115_transcriptome},\
${EPI116_transcriptome},\
${EPI123_transcriptome},\
${EPI124_transcriptome} %" \
"$maker_opts_file"
# Set proteomes to use
sed -i "/^protein=/ s% %\
${gigas_proteome},\
${panopea_td_proteome},\
${pgen_td_ctenidia_proteome},\
${pgen_td_gonad_proteome},\
${pgen_td_heart_proteome},\
${pgen_td_juv_EPI115_proteome},\
${pgen_td_juv_EPI116_proteome},\
${pgen_td_juv_EPI123_proteome},\
${pgen_td_juv_EPI124_proteome},\
${pgen_td_larvae_EPI99_proteome},\
${virginica_proteome} \
%" \
"$maker_opts_file"
# Set RepeatModeler library to use
sed -i "/^rmlib=/ s% %$repeat_library %" "$maker_opts_file"
sed -i "/^est_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.est2genome.gff %" maker_opts.ctl
sed -i "/^protein_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.protein2genome.gff %" maker_opts.ctl
sed -i "/^rm_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.repeats.gff %" maker_opts.ctl
sed -i "/^snaphmm=/ s% %Pgenerosa_v074_snap01.hmm %" maker_opts.ctl
fi
## Run Maker
### Set basename of files and specify number of CPUs to use
mpiexec -n 56 $maker \
-base Pgenerosa_v074_snap01
## Merge gffs
${gff3_merge} -d Pgenerosa_v074_snap01.maker.output/Pgenerosa_v074_snap01_master_datastore_index.log
## GFF with no FastA in footer
${gff3_merge} -n -s -d Pgenerosa_v074_snap01.maker.output/Pgenerosa_v074_snap01_master_datastore_index.log > Pgenerosa_v074_snap01.maker.all.noseqs.gff
## Run SNAP training, round 2
cd "${wd}"
cd snap02
${maker2zff} ../snap01/Pgenerosa_v074_snap01.all.gff
${fathom} -categorize 1000 genome.ann genome.dna
${fathom} -export 1000 -plus uni.ann uni.dna
${forge} export.ann export.dna
${hmmassembler} genome . > Pgenerosa_v074_snap02.hmm
## Initiate third and final Maker run.
if [ ! -e maker_opts.ctl ]; then
$maker -CTL
sed -i "/^genome=/ s% %$genome %" maker_opts.ctl
# Set transcriptomes to use
sed -i "/^est=/ s% %\
${ctendia_transcriptome},\
${gonad_transcriptome},\
${heart_transcriptome},\
${EPI99_transcriptome},\
${EPI115_transcriptome},\
${EPI116_transcriptome},\
${EPI123_transcriptome},\
${EPI124_transcriptome} %" \
"$maker_opts_file"
# Set proteomes to use
sed -i "/^protein=/ s% %\
${gigas_proteome},\
${panopea_td_proteome},\
${pgen_td_ctenidia_proteome},\
${pgen_td_gonad_proteome},\
${pgen_td_heart_proteome},\
${pgen_td_juv_EPI115_proteome},\
${pgen_td_juv_EPI116_proteome},\
${pgen_td_juv_EPI123_proteome},\
${pgen_td_juv_EPI124_proteome},\
${pgen_td_larvae_EPI99_proteome},\
${virginica_proteome} \
%" \
"$maker_opts_file"
# Set RepeatModeler library to use
sed -i "/^rmlib=/ s% %$repeat_library %" "$maker_opts_file"
sed -i "/^est_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.est2genome.gff %" maker_opts.ctl
sed -i "/^protein_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.protein2genome.gff %" maker_opts.ctl
sed -i "/^rm_gff=/ s% %../Pgenerosa_v074.maker.all.noseqs.repeats.gff %" maker_opts.ctl
sed -i "/^snaphmm=/ s% %Pgenerosa_v074_snap02.hmm %" maker_opts.ctl
fi
## Run Maker
### Set basename of files and specify number of CPUs to use
mpiexec -n 56 $maker \
-base Pgenerosa_v074_snap02
## Merge gffs
${gff3_merge} \
-d Pgenerosa_v074_snap02.maker.output/Pgenerosa_v074_snap02_master_datastore_index.log
## GFF with no FastA in footer
${gff3_merge} -n -s -d Pgenerosa_v074_snap02.maker.output/Pgenerosa_v074_snap02_master_datastore_index.log > Pgenerosa_v074_snap02.maker.all.noseqs.gff
## Merge FastAs
${fasta_merge} \
-d Pgenerosa_v074_snap02.maker.output/Pgenerosa_v074_snap02_master_datastore_index.log
# Create copies of files for mapping
cp "${maker_prot_fasta}" "${maker_prot_fasta_renamed}"
cp "${maker_transcripts_fasta}" "${maker_transcripts_fasta_renamed}"
cp "${snap02_gff}" "${snap02_gff_renamed}"
# Map IDs
## Change gene names
${map_ids} \
--prefix PGEN_ \
--justify 8 \
"${snap02_gff}" \
> "${id_map}"
## Map GFF IDs
${map_gff_ids} \
"${id_map}" \
"${snap02_gff_renamed}"
## Map FastAs
### Proteins
${map_fasta_ids} \
"${id_map}" \
"${maker_prot_fasta_renamed}"
### Transcripts
${map_fasta_ids} \
"${id_map}" \
"${maker_transcripts_fasta_renamed}"
# Run InterProScan 5
## disable-precalc since this requires external database access (which Mox does not allow)
cd "${ips_dir}"
${interproscan} \
--input "${maker_prot_fasta_renamed}" \
--goterms \
--output-file-base ${ips_base} \
--disable-precalc
# Run BLASTp
cd "${blastp_dir}"
${blastp} \
-query "${maker_prot_fasta_renamed}" \
-db ${sp_db_blastp} \
-out "${maker_blastp}" \
-max_target_seqs 1 \
-evalue 1e-6 \
-outfmt 6 \
-num_threads 28
# Functional annotations
cd "${wd}"
## Add putative gene functions
### GFF
${functional_gff} \
${sp_db_blastp} \
"${maker_blastp}" \
"${snap02_gff_renamed}" \
> ${put_func_gff}
### Proteins
${functional_fasta} \
${sp_db_blastp} \
"${maker_blastp}" \
"${maker_prot_fasta_renamed}" \
> ${put_func_prot}
### Transcripts
${functional_fasta} \
${sp_db_blastp} \
"${maker_blastp}" \
"${maker_transcripts_fasta_renamed}" \
> ${put_func_trans}
## Add InterProScan domain info
### Add searchable tags
${ipr_update_gff} \
${put_func_gff} \
"${ips_dir}"/${ips_name} \
> ${put_domain_gff}
### Add viewable features for genome browsers (JBrowse, Gbrowse, Web Apollo)
${iprscan2gff3} \
"${ips_dir}"/${ips_name} \
"${snap02_gff_renamed}" \
> ${ips_domains}RESULTS
Output folder:
Well, this ran relatively quickly: A little over seven days.
The important files:
- Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff
- GFF file with all contigs annotated with putative functions and functional domains.
- INCLUDES SEQUENCE FASTAS AT END OF FILE!
- Generated with one round of MAKER gene prediction, followed by two rounds of SNAP ab-initio gene prediction.
- MD5: 5a17f8dd40d534a544bec166c1b0c8be
- Pgenerosa_v074_genome_snap02.all.maker.proteins.renamed.putative_function.fasta (902KB)
- Annotated proteins FastA file.
- Generated with one round of MAKER gene prediction, followed by two rounds of SNAP ab-initio gene prediction.
- Pgenerosa_v074_genome_snap02.all.maker.transcripts.renamed.putative_function.fasta (2.5MB)
- Annotated transcripts FastA file.
- Generated with one round of MAKER gene prediction, followed by two rounds of SNAP ab-initio gene prediction.
I should’ve just split the GFF as part of the Mox SBATCH script, but I didn’t so I did it locally on my computer. Here are the commands for splitting the GFF. All the GFF files have been addeed to our Genomic Resources wiki (GitHub).
CDS GFF:
- Pgenerosa_v074.CDS.gff (1.3MB)
- MD5:
0585919b6d819efee8c86fb3bdc104b6
awk 'BEGIN { print "##gff-version 3" ; } $3 == "CDS" {print}' \
Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff \
> Pgenerosa_v074.CDS.gffexon GFF:
- Pgenerosa_v074.exon.gff (1.4MB)
- MD5:
92c1419894fb252d7c913313c725f3d5
awk 'BEGIN { print "##gff-version 3" ; } $3 == "exon" {print}' \
Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff \
> Pgenerosa_v074.exon.gffgene GFF:
- Pgenerosa_v074.gene.gff
- MD5:
8bf8bb60ce5fc42f2dfad2b5ae236abb
awk 'BEGIN { print "##gff-version 3" ; } $3 == "gene" {print}' \
Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff \
> Pgenerosa_v074.gene.gffmRNA GFF:
- Pgenerosa_v074.mRNA.gff
- MD5:
d1a6aba21b60f109510ee2399e03fb2c
awk 'BEGIN { print "##gff-version 3" ; } $3 == "mRNA" {print}' \
Pgenerosa_v074_genome_snap02.all.renamed.putative_function.domain_added.gff \
> Pgenerosa_v074.mRNA.gffWith all of that out of the way, a cursory glance at the results are, honestly, quite shocking. A quick grep -c ">" on the FastA files reveals:
- 1712 proteins/transcripts
This is compared to 53,035 proteins/transcripts identified/annotated in [the Pgenerosa_v070 annotation](https://robertslab.github.io/sams-notebook/posts/2019/2019-02-28-Genome-Annotation—Pgenerosa_v070-MAKER-on-Mox/transcripts in the Pgenerosa_v071 annotation (only contigs >10kbp).
Additionally, annotations only occur on 2 out of 18 scaffolds: - PGA_scaffold_17 - PGA_scaffold_18.
Surprisingly, these two scaffolds comprise two of the three shortest (35Mbp and 28Mbp) of the 18 scaffolds subjected to annotation.
So, what does this mean? A few thoughts/conjectures in no particular order or prioritization:
Limiting to the longest 18 scaffolds chosen to create this Pgenerosa_v074 subset was the wrong approach. Looking back at the GitHub issue where scaffold selection was discussed reveals the apparent confusion with which assembly provided by Phase Genomics is the proper assembly to use. However, that discussion seems to agree that the “manually curated” version of their genome assembly (which is what was used here) is the proper one to use.
Something went awry in the annotation. This will be difficult/impossible to assess, particularly since two of the scaffolds were successfully annotated. If two worked, why wouldn’t the other 16? All 18 scaffolds were presented to MAKER as a single multi-FastA file. Presumably, each individual FastA would be processed by MAKER in the same way.
The data is real and the majority of genes in the P.generosa genome lie outside of these large scaffolds. If this is the case, this means that there are lengthy stretches of DNA that do not have readily-identifiable genomic features. It also suggests that, overall, the assembly we have (Pgenerosa_v070) is quite fragmented.
So, to assess some of this (particularly points 2 and 3 above), I decided to look at the three annotations we have for P.generosa using IGV on some of the 18 scaffolds that are present in each of the three previous assembly variations we’ve annotated with MAKER.
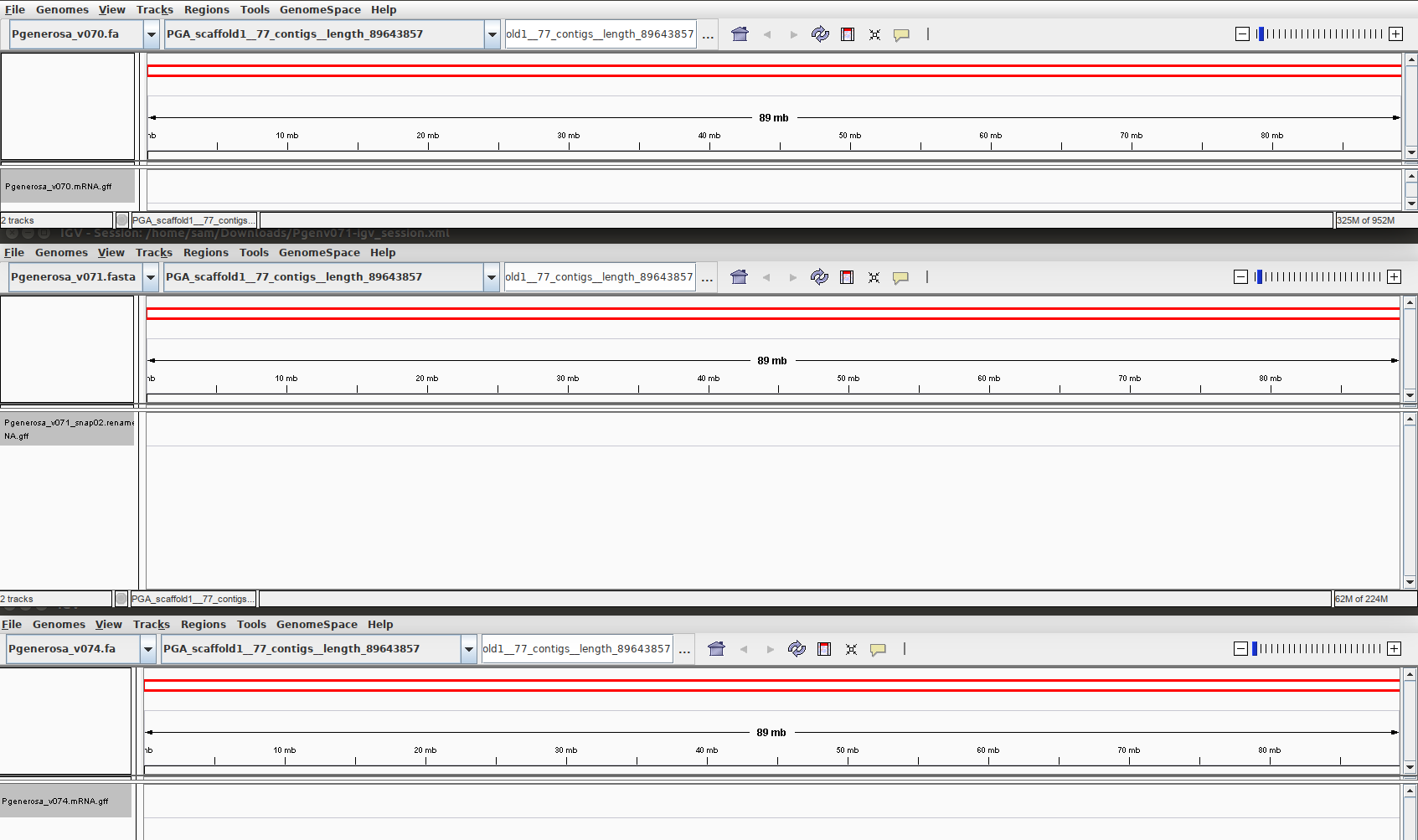
PGA_scaffold_1
No annotations found by any of the three different annotations. Surprising, as this is the single longest scaffold in each of the three assemblies (89 Mbp)
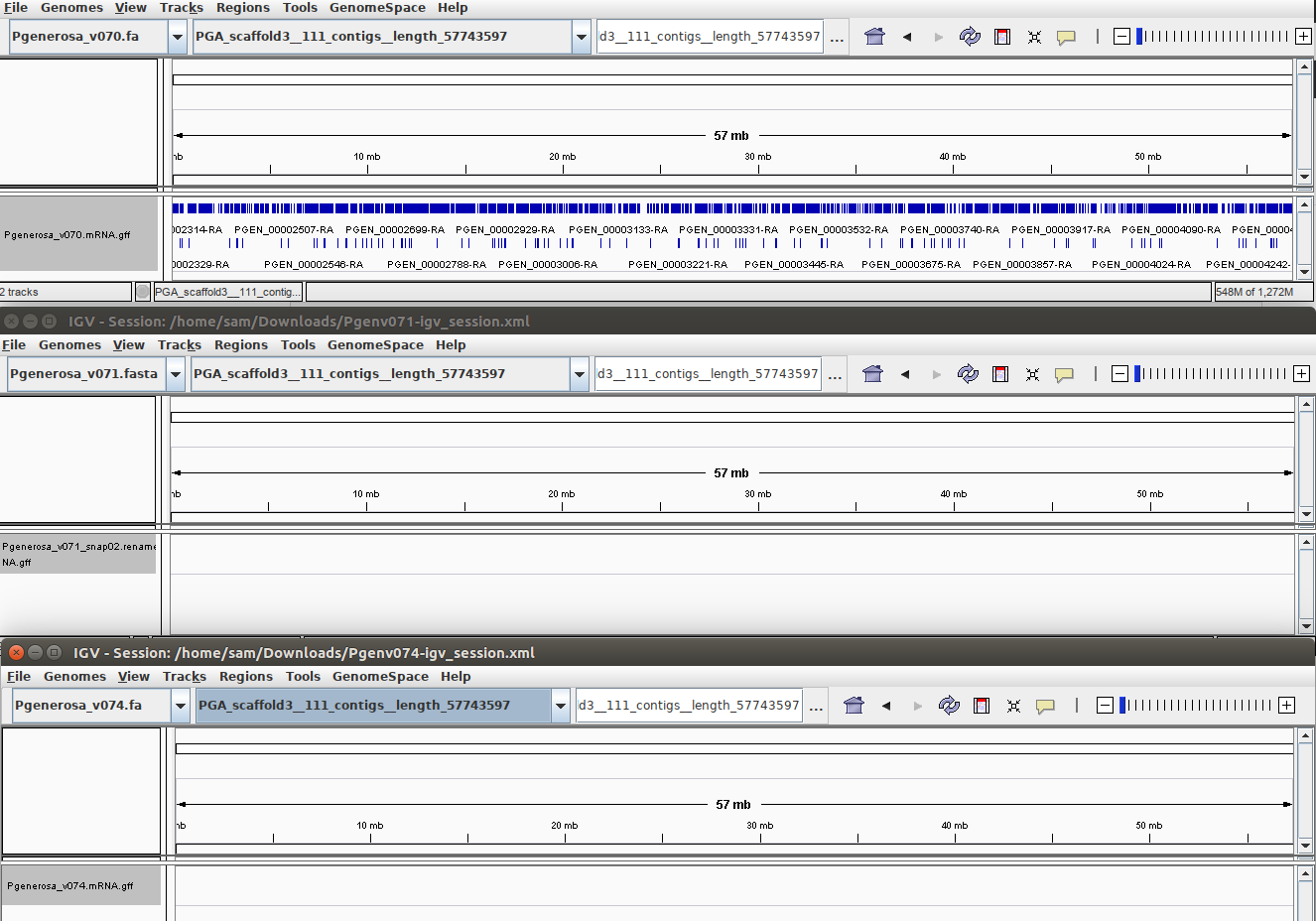
PGA_scaffold_3
Annotations only appear in the Pgenerosa_v070 assembly.
PGA_scaffold_18
Annotations are found in each of the three assemblies.

Yeesh, so what do those IGV alignments tell us?
The single longest scaffold appears to have no identifiable genomic features, as no annotations are present in any of the various assemblies. That’s a pretty lengthy stretch of DNA to not have anything there. Caveat: haven’t looked at transposable elements, nor other repeats. Is it possible that an 89Mbp stretch of DNA could be comprised solely of low-complexity sequence?
The annotation process appears to be inconsistent. This is troublesome. Although this comes with a catch: I did not use the exact same data for each of the three annotations. For example, the Pgenerosa_v070 and Pgenerosa_v071 annotations used the singular P.generosa transcriptome assembly (from 20180827)for EST evidence, whereas the Pgenerosa_v074 annotation utilized tissue-specific transcriptome assemblies (from 20190409) and Transdecoder peptide data from each of these assemblies. Could these changes account for the differences we see? Oddly, and this thought is solely based on the limited IGV comparisons posted above, it seems like the fewer the overall number of sequences in an assembly, the fewer annotations MAKER is able to generate within any given scaffold.
Now what?
Well, I have the Pgenerosa_v074 assembly currently running on the Genome Sequence Annotation Server (GenSAS) using most of the same files that I used for MAKER on Mox (exceptions are repeat masking/modeling files - those steps will be run on GenSAS). It will be interesting to see how the MAKER and GenSAS annotations compare.
I could also re-run the Pgenerosa_v071 annotation using the exact same files as Pgenerosa_v074 on GenSAS, as the files I need are already on their servers and it will be trivial to initiate the annotation process…