Steven noticed that the M-Bias plots generated by Bismark from these files was a little wonky and asked that I try trimming them a bit more. The files were originally quality/adaptor trimmed with TrimGalore! on 20180516.
I decided to use some trimming software called fastp, since it had a lot of options (including trimming without the need for quality filtering/trimming, as well as being multi-threaded).
Looking at the M-Bias plots and the original FastQC assessment, I opted to trim the first 20bp from the 5’ end of all reads.
I followed this up with FastQC and MultiQC.
Use the following Bash script to initiate file transfer to Mox and call the SBATCH script:
#!/bin/bash
## Rsync P.generosa EPI files and then run SBATCH script for hard trimming.
rsync -av --progress owl:/volume1/web/Athaliana/20180516_geoduck_trimgalore_rrbs/*.fq.gz .
sbatch 20190923_pgen_fastp_EPI_trimming.shSBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=pgen_fastp_trimming_EPI
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=10-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20190923_pgen_fastp_EPI_trimming
# This script is called by 20190923_pgen_EPI_rsync.sh. That script transfers the FastQ files
# to the working directory from: https://owl.fish.washington.edu/Athaliana/20180516_geoduck_trimgalore_rrbs
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
# Set number of CPUs to use
threads=28
# Set number of nucleotides to hard trim
num_nucs_trim=20
# Paths to programs
fastp=/gscratch/srlab/programs/fastp-0.20.0/fastp
## Inititalize arrays
fastq_array_R1=()
fastq_array_R2=()
R1_names_array=()
R2_names_array=()
# Create array of fastq R1 files
for fastq in *R1*.gz
do
fastq_array_R1+=("${fastq}")
done
# Create array of fastq R2 files
for fastq in *R2*.gz
do
fastq_array_R2+=("${fastq}")
done
# Create array of sample names
## Uses awk to parse out sample name from filename
for R1_fastq in *R1*.gz
do
R1_names_array+=($(echo "${R1_fastq}" | awk -F"." '{print $1}'))
done
# Create array of sample names
## Uses awk to parse out sample name from filename
for R2_fastq in *R2*.gz
do
R2_names_array+=($(echo "${R2_fastq}" | awk -F"." '{print $1}'))
done
# Create list of fastq files used in analysis
## Uses parameter substitution to strip leading path from filename
for fastq in *.gz
do
echo "${fastq}" >> fastq.list.txt
done
# Run fastp on files and trim set number of nucleotides from 5' end of reads
for index in "${!fastq_array_R1[@]}"
do
R1_sample_name=$(echo "${R1_names_array[index]}")
R2_sample_name=$(echo "${R2_names_array[index]}")
${fastp} \
--in1 "${fastq_array_R1[index]}" \
--in2 "${fastq_array_R2[index]}" \
--disable_quality_filtering \
--disable_length_filtering \
--disable_adapter_trimming \
--trim_front1 ${num_nucs_trim} \
--trim_front2 ${num_nucs_trim} \
--thread ${threads} \
--out1 "${R1_sample_name}".20bp-trim.fq.gz \
--out2 "${R2_sample_name}".20bp-trim.fq.gz
# Remove original FastQ files
rm "${fastq_array_R1[index]}" "${fastq_array_R2[index]}"
doneRESULTS
This was pretty quick, ~2.75hrs:

Output folder:
fastp report (HTML):
MultiQC report (HTML):
Firstly, it turns out that fastp is pretty awesome! It automatically generates a summary HTML file with before and after trimming data, similar to that of FastQC. Had I realized this, I might not have bothered with FastQC…
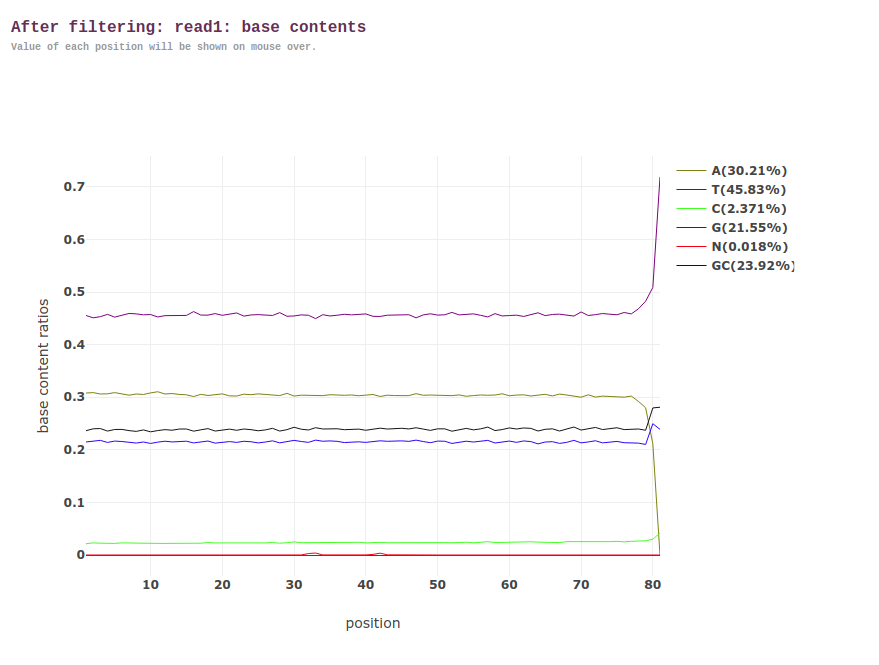
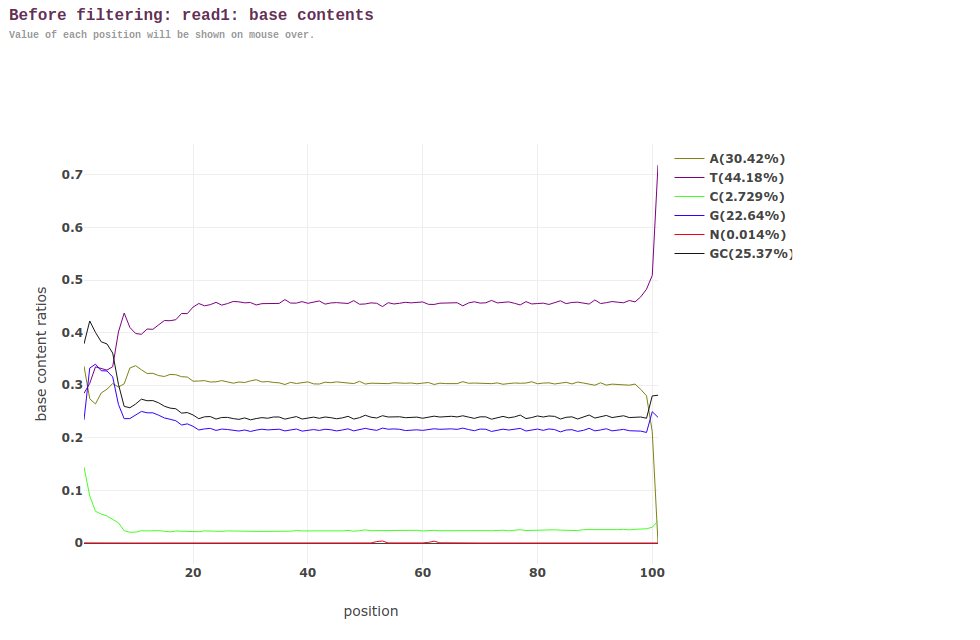
Anyway, with that being said, this is how the before/after look for Read 1s via the fastp HTML:
BEFORE
AFTER