After receiving the rest of the crab data and concatenating it all together, I ran FastQC and MultiQC on the FastQ files.
RESULTS
Output folder:
MultiQC Report (HTML):
So, that’s done. However, I’ve noticed that one of the samples (sample ID 329775) only has ~42M reads (circled in red below):
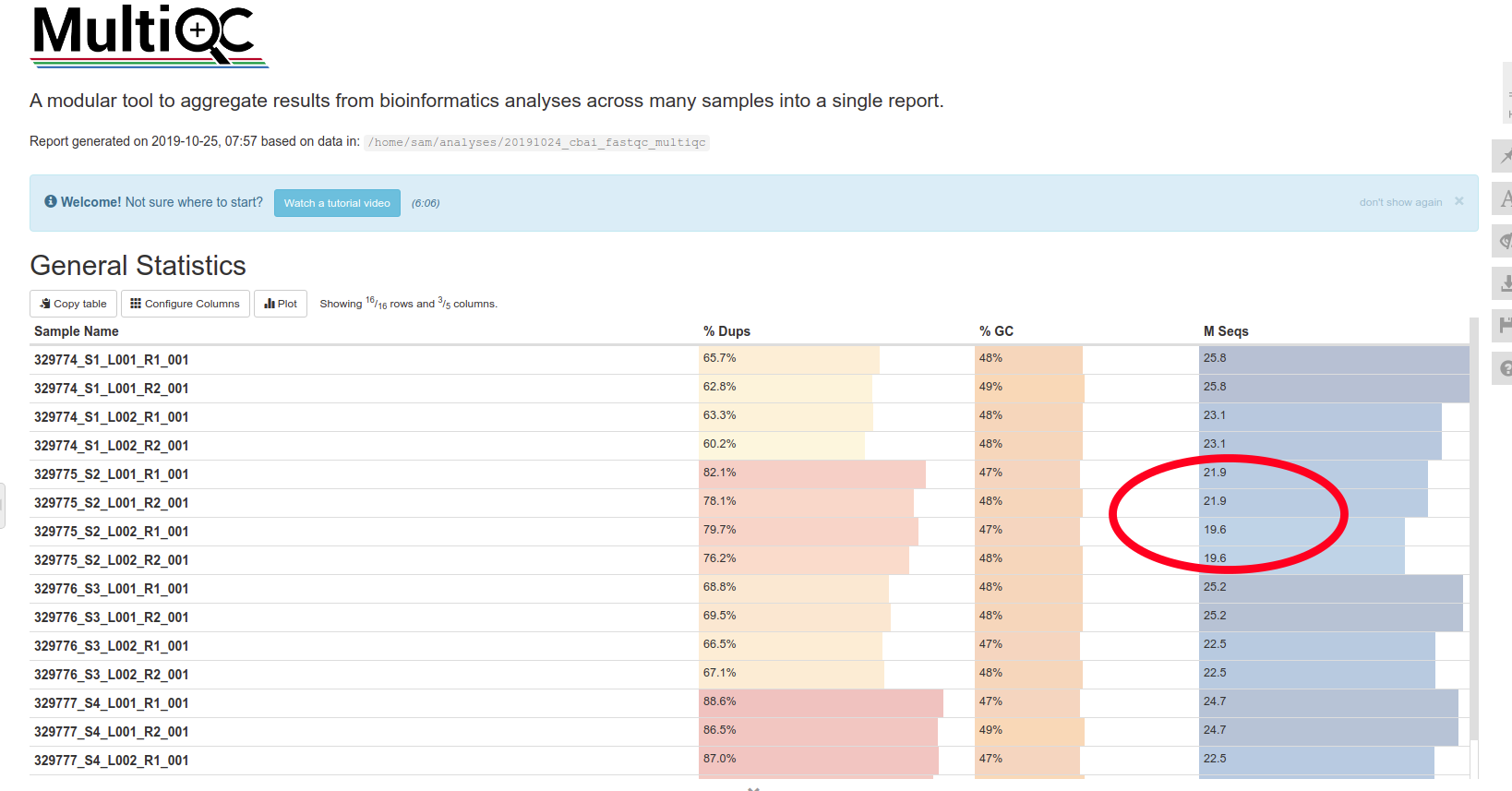
This read count is ~16% less than what we were quoted for. Unfortunately, the quote was for “~50M reads per sample”. That “~” leads to a fair amount of ambiguity. The rest of the samples hover around 5 - 6% less than the 50M read mark. Is that acceptable? I don’t know. In hindsight, I should’ve clarified what they meant on the quote. Of course, the other option is that this facility (Northwest Genomics Center)get their act together and write quotes that aren’t ambiguous (e.g. promise >= 50M reads; then there’s absolutely no confusion about what the customer is supposed to receive)!!
UPDATE
After emailing the sequencing facility, it turns out the read count “issue” is a difference in the terminology. They’re reporting absolute number of reads generated (I agree with this, btw). So, we have most certainly received >50M for each sample. The confusion was related to the way other facilities refer to read counts. Most other facilities will count a read pair (e.g. R1 an R2) as a single read.
Regardless, I still think they need to do away with ambiguity when quoting projects. :)