I performed the initial Lambda sequencing test on 20200107 and everything went smoothly, so I’m ready to give the NanoPore (ONT) MinION a run with an actual sample!
I isolated gDNA from an uninfected C.bairdi muscle sample yesterday (sample 20102558-2729). Earlier today I ran that DNA on a gel to assess it’s quality/integrity and it didn’t look very good - pretty degraded.
Despite that fact, I’ll run this on the NanoPore MinION. This will provide me with additional experience on using the system and will also provide us with info about using degraded DNA. Presumably, sequencing will proceed without issue and we’ll just end up with shorter read lengths than if we had higher quality input DNA.
The sample DNA was prepared according to the protocol for the Rapid Sequencing Kit (SQK-RAD004) and run on a FLO-MIN106 (ID: FAL58500) flowcell. Data acquisition was set to run without basecalling for a period of 72hrs. This will make sure the raw Fast5 output files will be preserved (not sure if they’re saved or not when basecalling is enabled), but will require conversion to FastQ at a later date.
Start of the run was good, with 1226 pores available for sequencing (minimum for a “good” flowcell, per ONT, is 800 pores). The number of available pores dropped below 800 after ~3hrs, which doesn’t seem good. Based on the various metrics for monitoring a sequencing run, I decided to terminate this run after ~17hrs. Available pores had dropped to 270 and data acquisition was minimal. With that being said, I should be able to wash the flowcell to restore everything back to normal (i.e. clean out clogged pores) and use it again.
I washed the flowcell according the protocol for the Flowcell Wash Kit (WFC_9088) and decided to do a second run with this same gDNA sample to see how well washing/reusing actually seems to work.
Prepared a fresh library as before and ran as before on the FLO-MIN106 (ID: FAL58500) flowcell. This time, I decided to just go with a “set it and forget it” approach. I set the run time to 72hrs and let the program finish. The washing/reusing of the flowcell didn’t really work as expected, though. Starting number of available pores was only 414, far short of the minimum of 800 indicative of a good flowcell. I’ll contact ONT about this and see what they can do.
RESULTS
Output folder:
First run outputs:
Fast5 directory:
Run report (PDF):


Second run outputs:
Fast5 directory:
Run report (PDF):
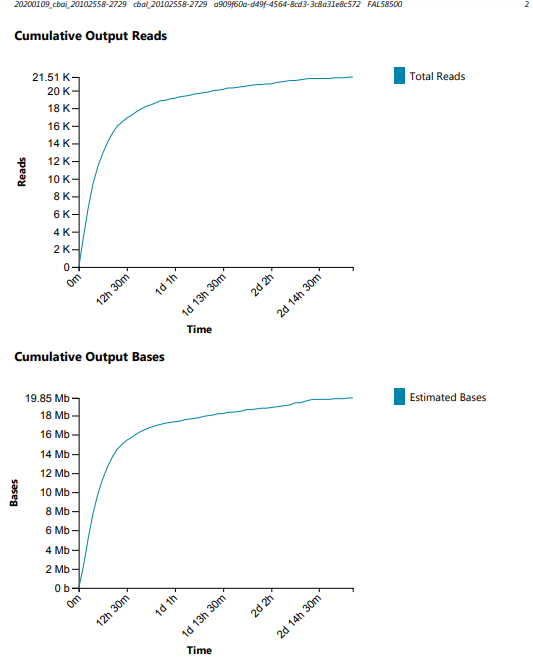
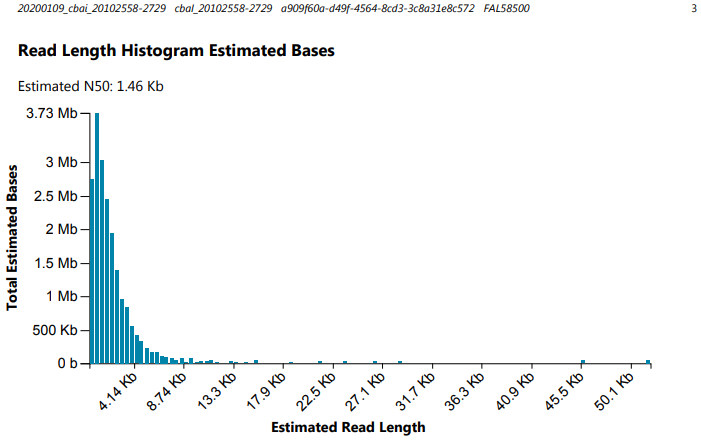
Alrighty, here’s how I interpret things.
First run:
~3x more reads
~3x more bases
Lower N50 (1.29Kbp vs 1.46Kbp)
Run time was 4.5x shorter
Everything’s as expected, except maybe the N50, and demonstrates the importance of the available sequencing pores in data acquisition.
Next up, convert the raw Fast5 files to FastQ.