I previously ran BLASTx and “meganized” the output DAA files on 20200103 (for reference, these include RNAseq data using a newly established “shorthand”: 2018, 2019) and now need to use MEGAN6 to bin the results into the proper taxonomies. This is accomplished using the MEGAN6 graphical user interface (GUI). This is how the process goes:
File > Import from BLAST…
Select all “meganized” DAA files for a given set of sequencing (e.g.
304428_S1_L001_R1_001.blastx.daa,304428_S1_L001_R2_001.blastx.daa,304428_S1_L002_R1_001.blastx.daa,304428_S1_L001_R2_001.blastx.daa)Check the “Paired Reads” box. (I don’t think this actually does anything, though…)
Click “Next”
Check the “Analyze Taxonomy Content” box.
Click “Load Accession mapping file” and find the mapping file used for “meganizing the DAA file”:
prot_acc2tax-Jul2019X1.abinClick “Next”
Click “Load Accession mapping file” and find the mapping file used for “meganizing the DAA file”:
acc2eggnog-Jul2019X.abinClick “Next”
Click “Load Accession mapping file” and find the mapping file used for “meganizing the DAA file”:
acc2interpro-Jul2019X.abinClick “Next” twice, to advance to the “SEED” tab.
Click “Load Accession mapping file” and find the mapping file used for “meganizing the DAA file”:
acc2seed-May2015XX.abinClick “Next” twice, to advance to the “LCA Params” tab.
Click “Apply”
This will initiate the import process and will create a special MEGAN file: RMA6.
NOTE: This will take a long time and will require a significant amount of disk space! The final files aren’t ridiculously large, but the intermediate file that gets generated quickly becomes extremely large (i.e. hundreds of GB)!
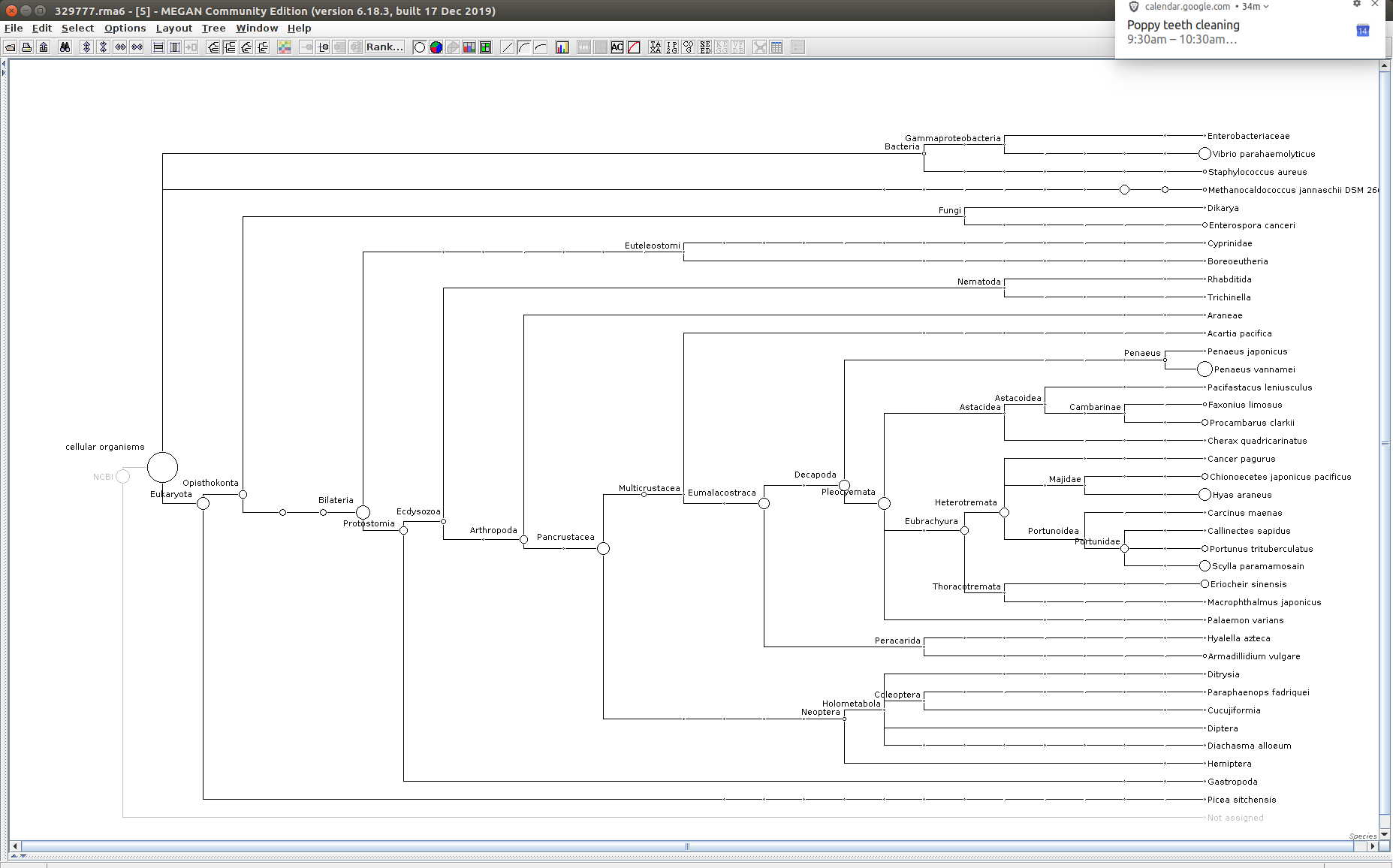
After that has completed, use the MEGAN6 GUI to “Open” the RMA6 file. Once the file loads, you will get a nice looking taxonomic tree! From here, you can select any part of the taxonomic tree by right-clicking on the desired taxonomy and “Extract reads…”. Here, you have the option to include “Summarized reads”. This option allows you to extract just the reads that are part of the exact classification you’ve selected or all those within and “below” the classification you’ve selected (i.e. summarized reads).
Extracted reads will be generated as FastA files.
Example:
If you select Arthropoda and do not check the box for “Summarized Reads” you will only get reads classified as Arthropoda! You will not get any reads with more specific taxonomies. However, if you select Arthropoda and you do check the box for “Summarized Reads”, you will get all reads classified as Arthropoda AND all reads in more specific taxonomic classifications, down to the species level.
I will extract reads from two phyla:
Arthropoda (for crabs)
Alveolata (for Hematodinium)
RESULTS
I put the RMA6 files in the original DIAMOND BLASTx/meganization folder from 20200103, as it seemed to make most sense organizational-wise to keep those together.
Output folder:
I put the extracted reads (FastA) here:
One good thing to see was that the samples that were considered “uninfected” (based on PCR/qPCR data) had no reads classified as Alveolata (see samples 329775 and 329777 below).
Taxonomic Trees
304428
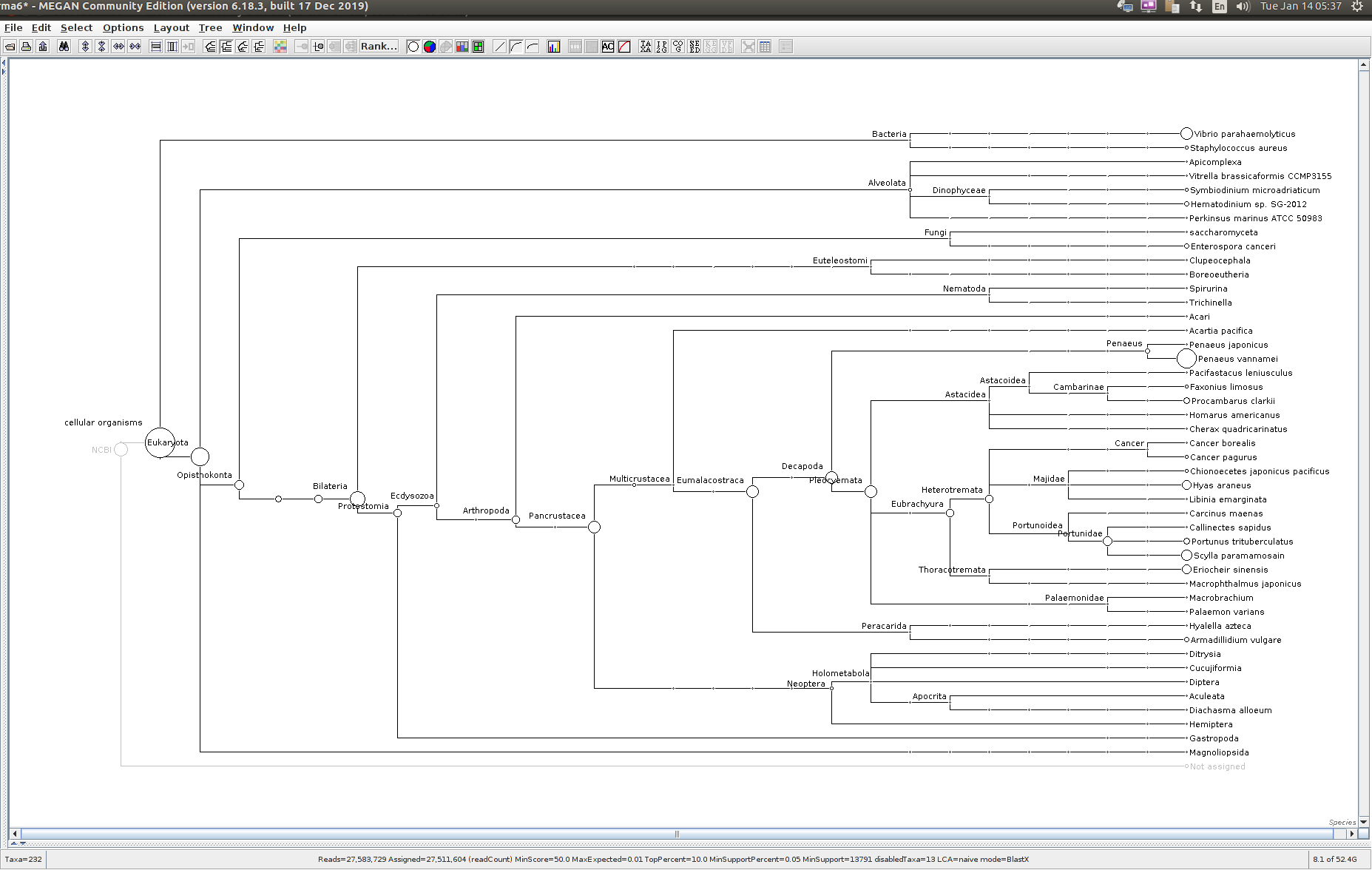
329774
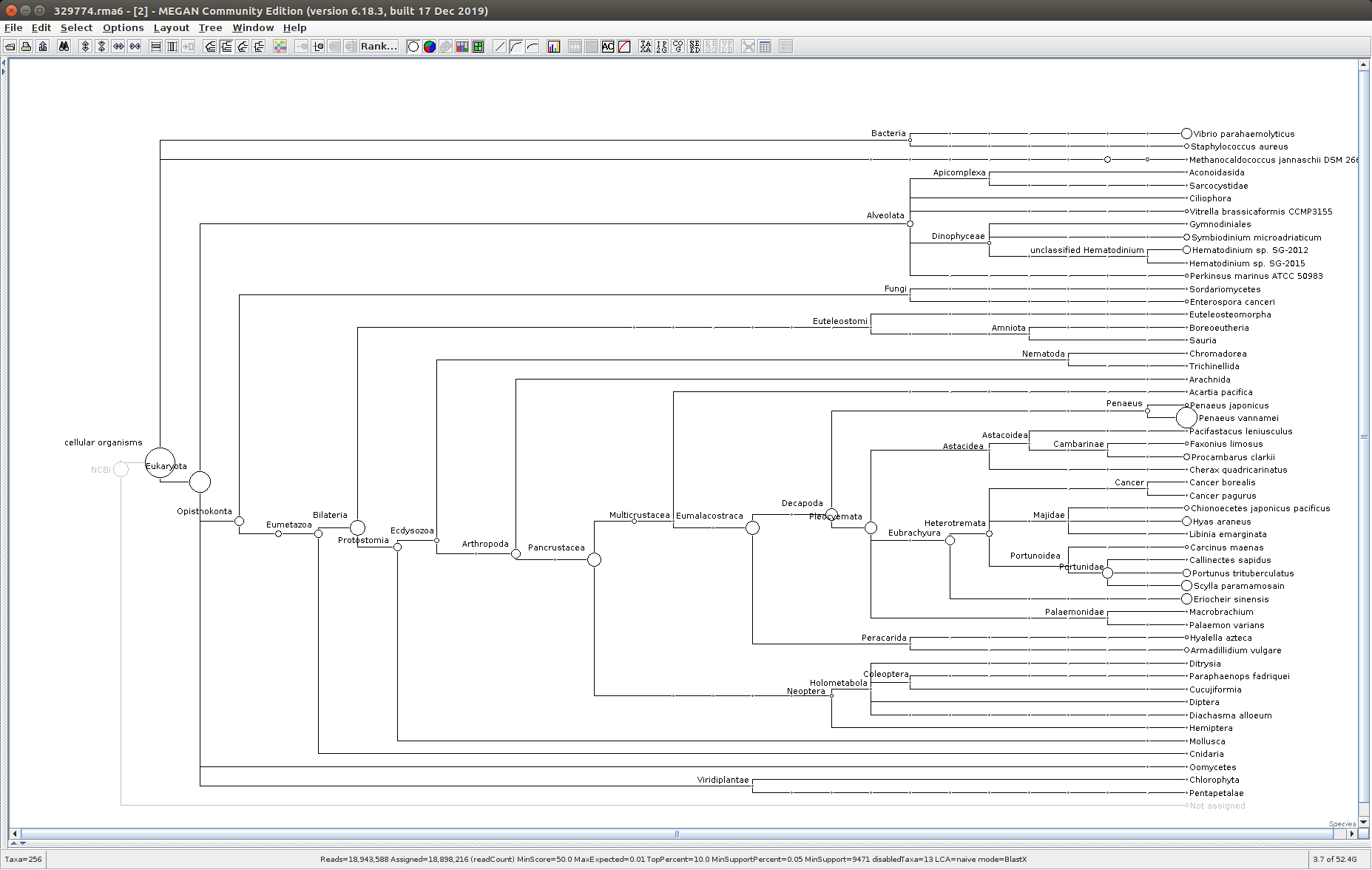
329775
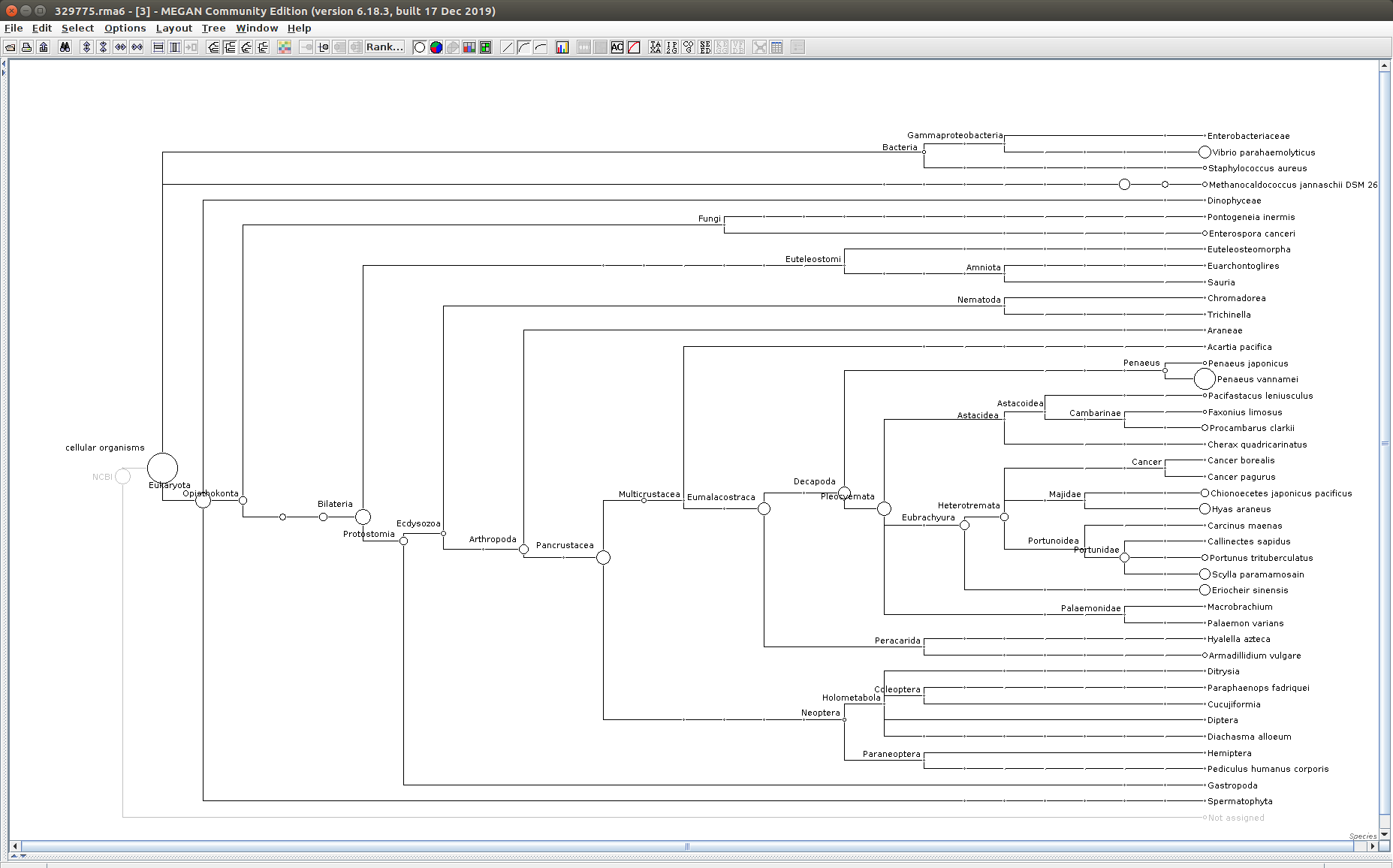
329776
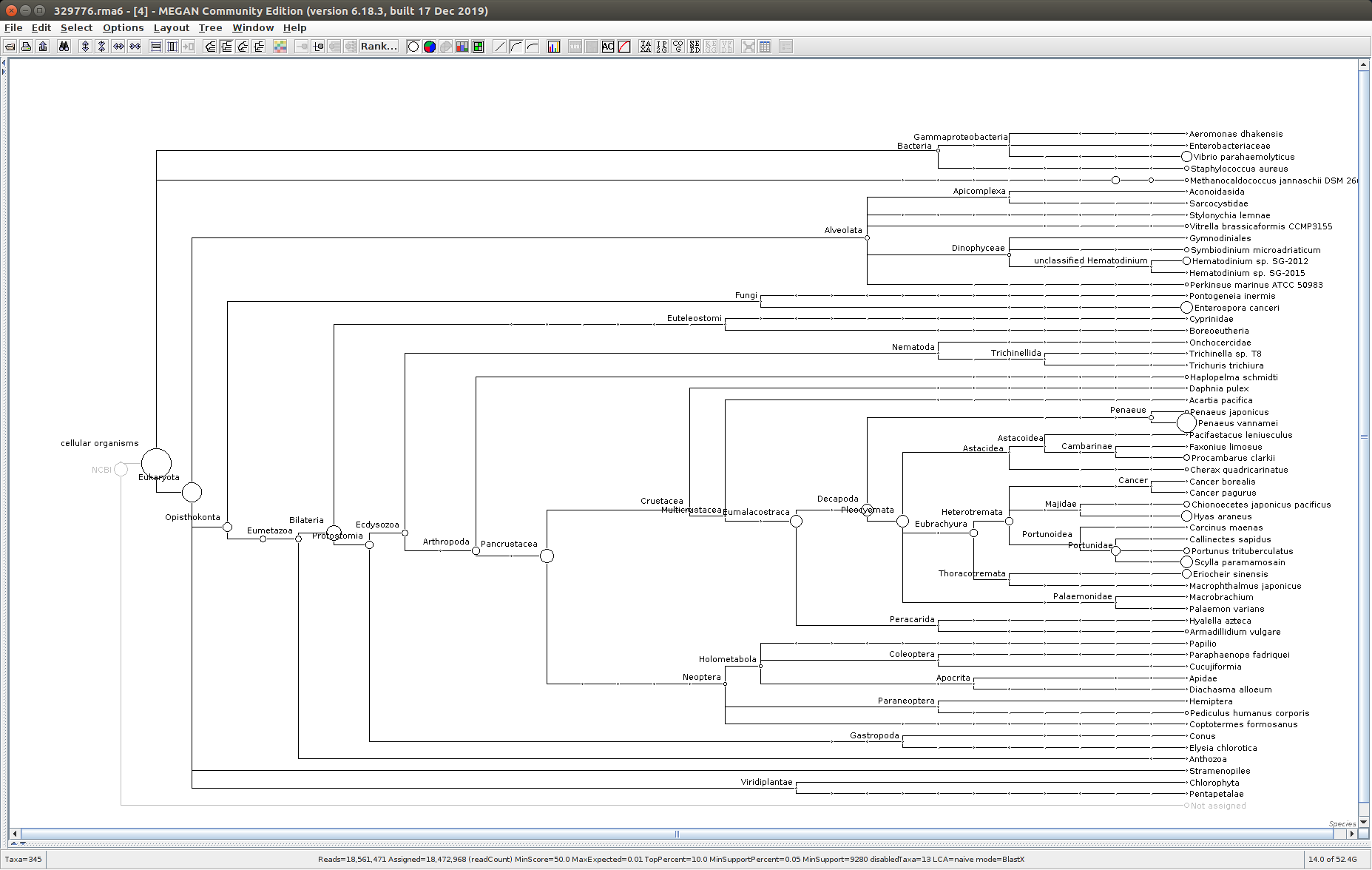
329777