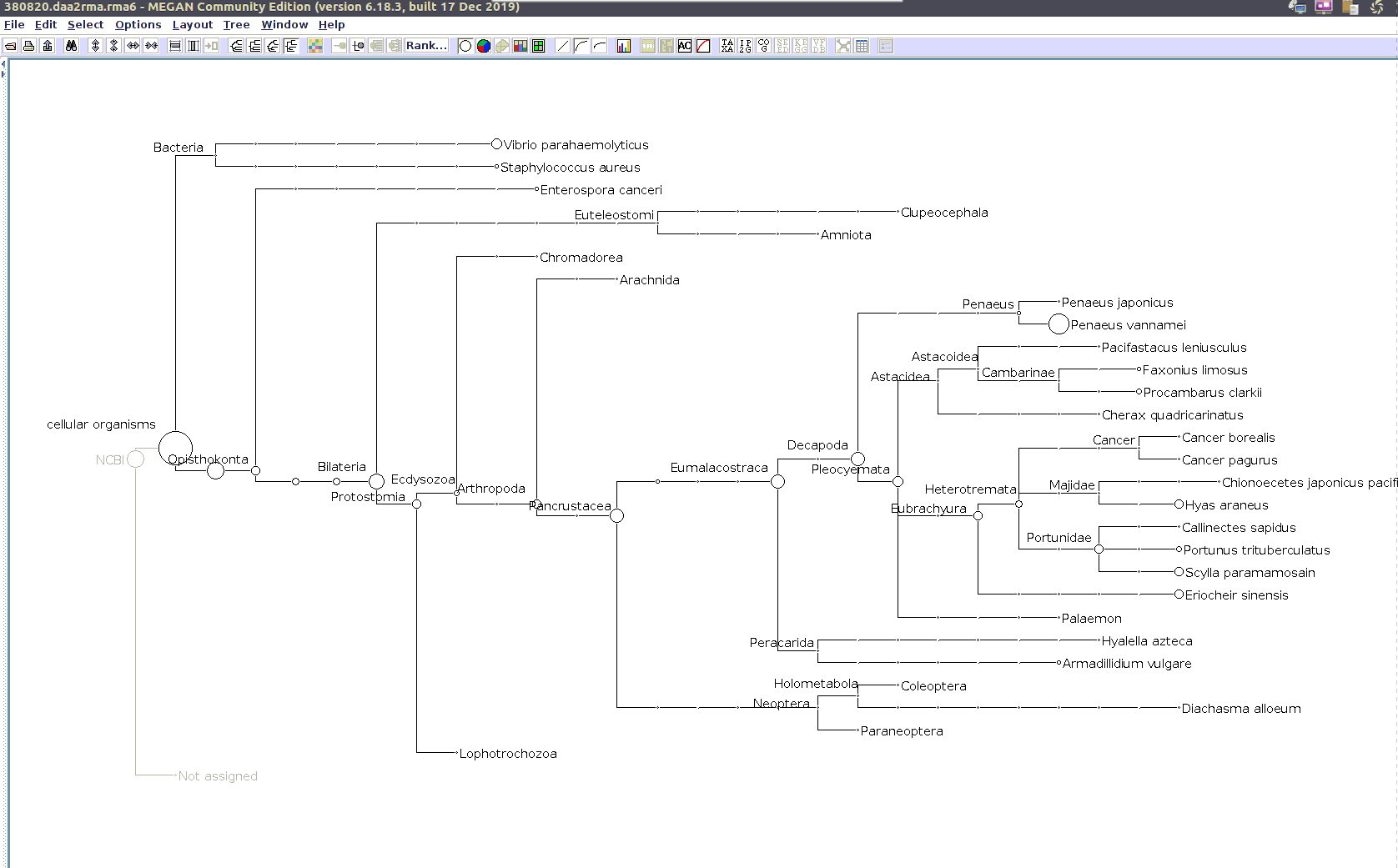
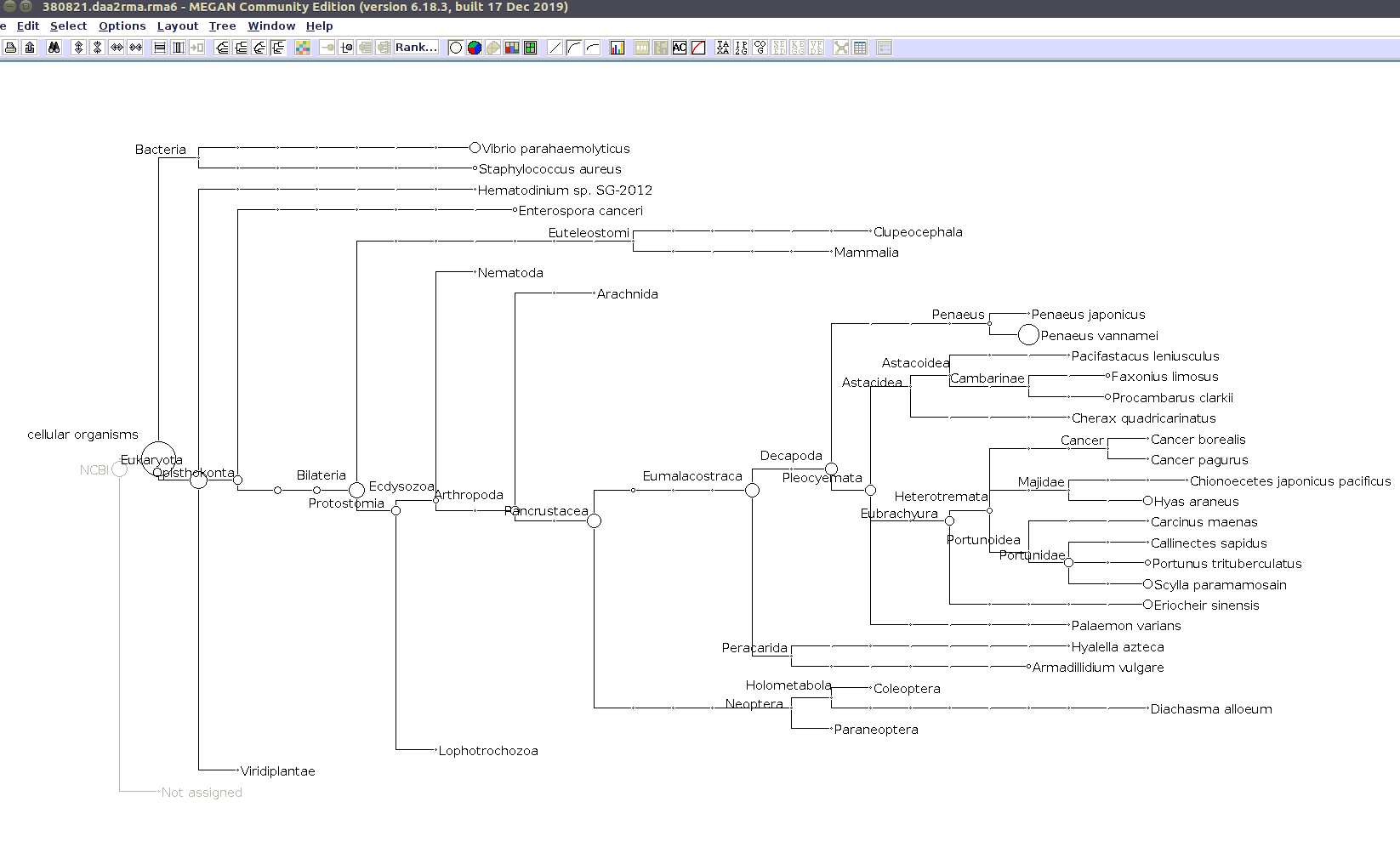
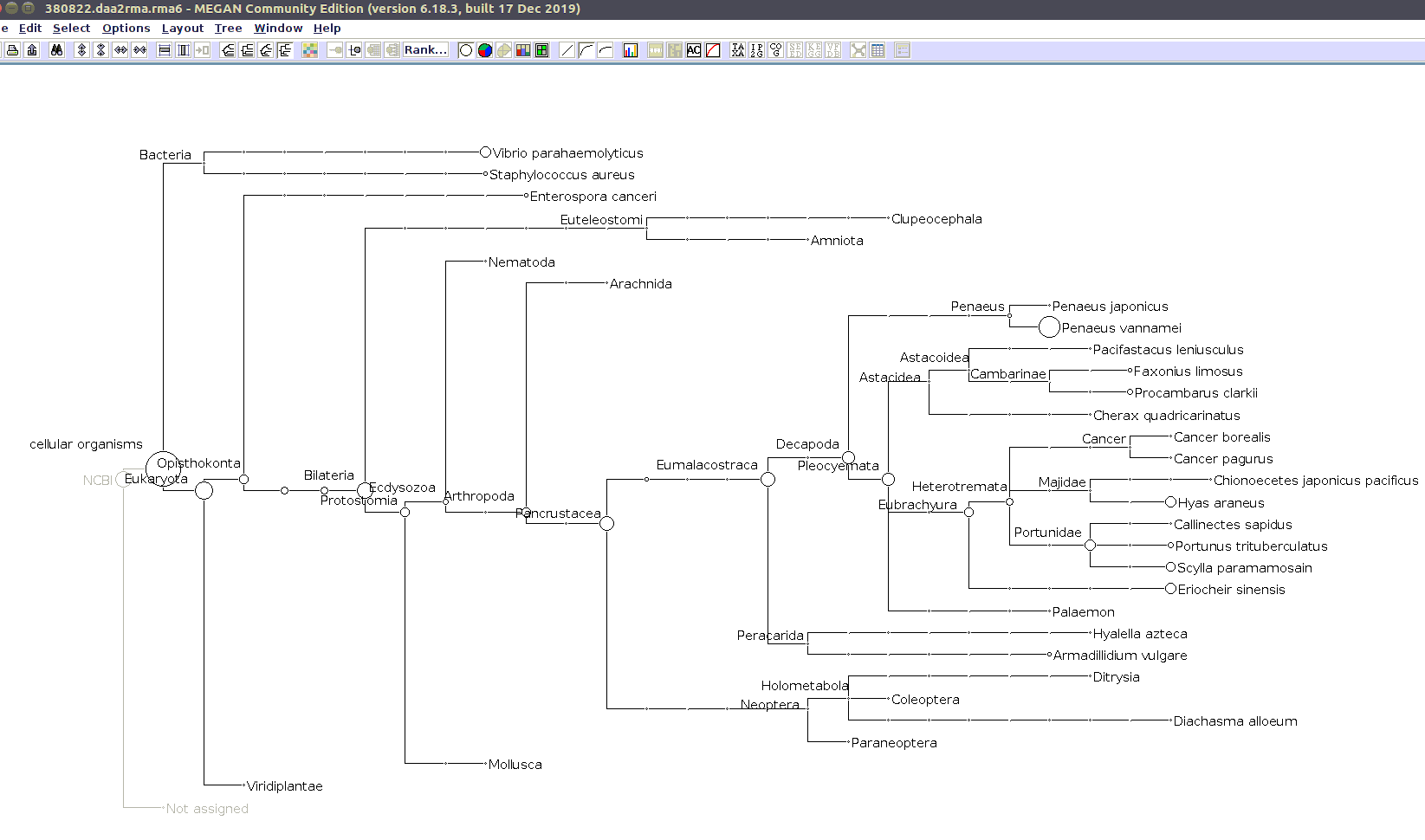
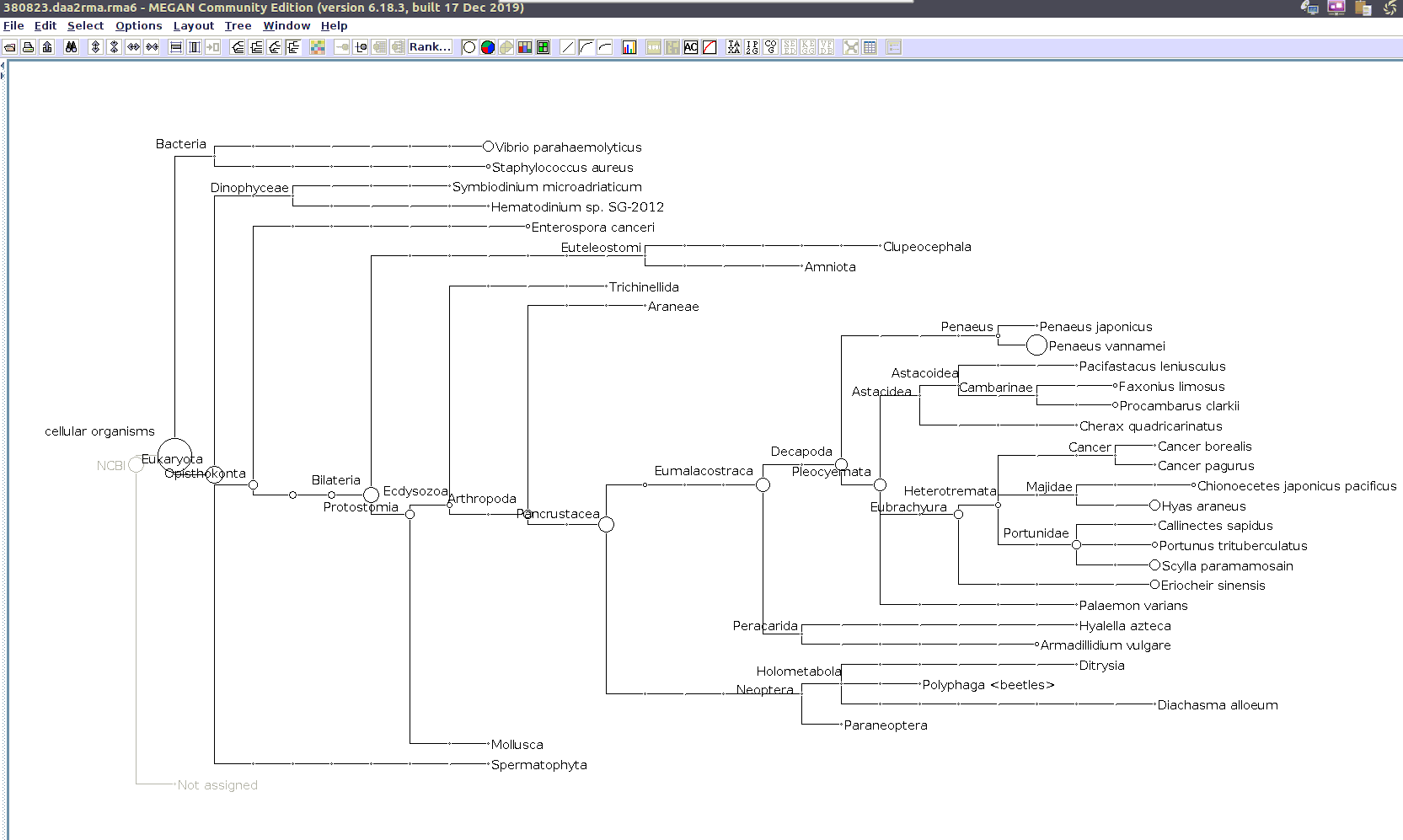
I previously annotated reads and converted them to the MEGAN6 format RMA6 on 20200414.
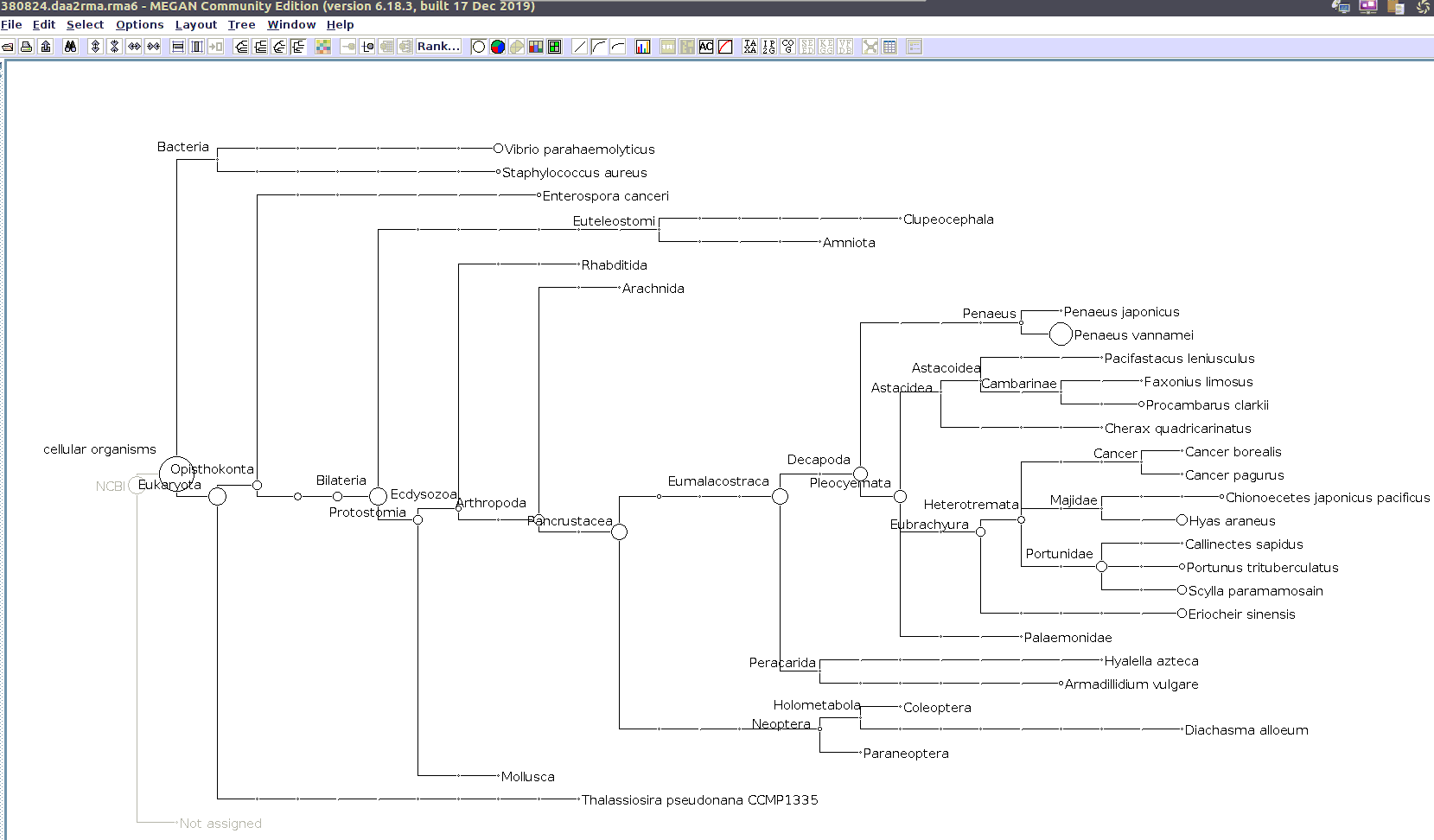
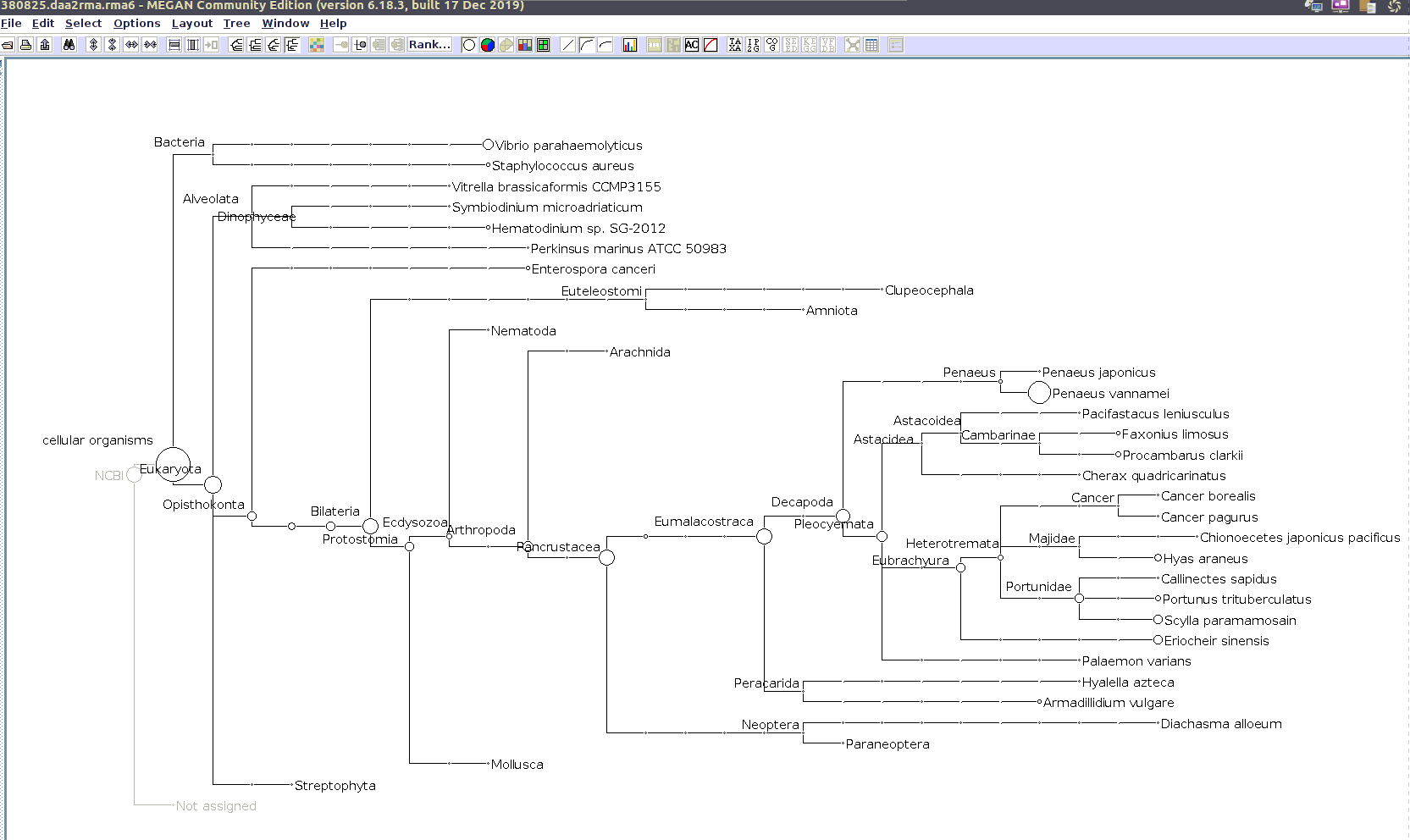
I’ll use the MEGAN6 GUI to “Open” the RMA6 file. Once the file loads, you get a nice looking taxonomic tree! From here, you can select any part of the taxonomic tree by right-clicking on the desired taxonomy and “Extract reads…”. Here, you have the option to include “Summarized reads”. This option allows you to extract just the reads that are part of the exact classification you’ve selected or all those within and “below” the classification you’ve selected (i.e. summarized reads).
Extracted reads will be generated as FastA files.
Example:
If you select Arthropoda and do not check the box for “Summarized Reads” you will only get reads classified as Arthropoda! You will not get any reads with more specific taxonomies. However, if you select Arthropoda and you do check the box for “Summarized Reads”, you will get all reads classified as Arthropoda AND all reads in more specific taxonomic classifications, down to the species level.
I will extract reads from two phyla:
Arthropoda (for crabs)
Alveolata (for Hematodinium)
After read extractions using MEGAN6, I’ll need to extract the actual reads from the trimmed FastQ files from 20200414.
It’s a bit convoluted, but I realized that the FastA headers were incomplete and did not distinguish between paired reads. Here’s an example:
R1 FastQ header:
@A00147:37:HG2WLDMXX:1:1101:5303:1000 1:N:0:AGGCGAAG+AGGCGAAG
R2 FastQ header:
@A00147:37:HG2WLDMXX:1:1101:5303:1000 2:N:0:AGGCGAAG+AGGCGAAG
However, the reads extracted via MEGAN have FastA headers like this:
>A00147:37:HG2WLDMXX:1:1101:5303:1000
SEQUENCE1
>A00147:37:HG2WLDMXX:1:1101:5303:1000
SEQUENCE2Those are a set of paired reads, but there’s no way to distinguish between R1/R2. This may not be an issue, but I’m not sure how downstream programs (i.e. Trinity) will handle duplicate FastA IDs as inputs. To avoid any headaches, I’ve decided to parse out the corresponding FastQ reads which have the full header info.
Here’s a brief rundown of the approach:
Create list of unique read headers from MEGAN6 FastA files.
Use list with
seqtkprogram to pull out corresponding FastQ reads from the trimmed FastQ R1 and R2 files.
This aspect of read extractions/concatenations is documented in the following Jupyter notebook (GitHub):
RESULTS
OUTPUT FOLDERS
Initial reads extracted as FastAs:
FastQ C.bairdi read extractions:
FastQ Hematodinium read extractions:
Taxonomic Trees