We’ve produced a number of C.bairid transcriptomes utilizing different assembly approaches (e.g. Arthropoda reads only, stranded libraries only, mixed strandedness libraries, etc) and we want to determine which of them is “best”. Trinity has a nice list of tools to assess the quality of transcriptome assemblies, but most of the tools rely on comparison to a transcriptome of a related species.
I was unable to readily find a crab transcriptome assembly anywhere, so decided to make our own assembly using RNAseq data from NCBI SRA BioProject PRJNA597187. This is from the Japanese blue crab, Portunus trituberculatus and has a fair amount of sequencing data, which should result in a pretty comprehensive transcriptome.
Before assembling, however, I want to determine if the libraries in this particular project were stranded or not, as Trinity has an option to indicate stranded libraries for use in the assembly. Trinity also offers a script to determine library strandedness which produces a set of violin plots to help discern the type of library one is working with.
Basically, you have to create a transcriptome assembly and then map the reads back to the assembly. Then, the Trinity script examine_strand_specificity.pl will generate the violin plots. I ran the Trinity script on just a single set of paired-end reads FastQs to make this happen relatively quickly. This was run on Mox.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=trinity_ptri_strand_check
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=9-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20200521_ptri_trinity_strandedness_check
### De novo transcriptome assembly of Portunus trituberculatus (Japanese blue crab)
### RNAseq data from NCBI BioProject PRJNA597187.
### Use single set of FastQ reads to determine library standedness.
# Exit script if a command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
# User-defined variables
reads_dir=/gscratch/srlab/sam/data/P_trituberculatus/RNAseq
threads=28
assembly_stats=assembly_stats.txt
# Paths to programs
trinity_dir="/gscratch/srlab/programs/trinityrnaseq-v2.9.0"
samtools="/gscratch/srlab/programs/samtools-1.10/samtools"
trinity_bowtie="${trinity_dir}/util/misc/run_bowtie2.pl"
trinity_strand="${trinity_dir}/util/misc/examine_strand_specificity.pl"
## Inititalize arrays
R1_array=()
R2_array=()
# Variables for R1/R2 lists
R1_list=""
R2_list=""
# Create array of fastq R1 files
R1_array=("${reads_dir}"/SRR10757128.sra_1.fastq)
# Create array of fastq R2 files
R2_array=("${reads_dir}"/SRR10757128.sra_2.fastq)
# Create list of fastq files used in analysis
## Uses parameter substitution to strip leading path from filename
for fastq in "${!R1_array[@]}"
do
{
echo "${R1_array[${fastq}]##*/}"
echo "${R2_array[${fastq}]##*/}"
} >> fastq.list.txt
done
# Create comma-separated lists of FastQ reads
R1_list=$(echo "${R1_array[@]}" | tr " " ",")
R2_list=$(echo "${R2_array[@]}" | tr " " ",")
# Run Trinity
## Not running as "stranded", due to mix of library types
${trinity_dir}/Trinity \
--seqType fq \
--max_memory 100G \
--CPU ${threads} \
--left "${R1_list}" \
--right "${R2_list}"
# Assembly stats
${trinity_dir}/util/TrinityStats.pl trinity_out_dir/Trinity.fasta \
> ${assembly_stats}
# Create FastA index
${samtools} faidx \
trinity_out_dir/Trinity.fasta
# Align reads to assembly
${trinity_bowtie} \
--target trinity_out_dir/Trinity.fasta \
--left "${R1_list}" \
--right "${R2_list}" \
| ${samtools} view \
--threads ${threads} \
-Sb - \
| ${samtools} sort \
--threads ${threads} \
- -o bowtie2.coordSorted.bam
# Examine strand specificity
${trinity_strand} bowtie2.coordSorted.bamRESULTS
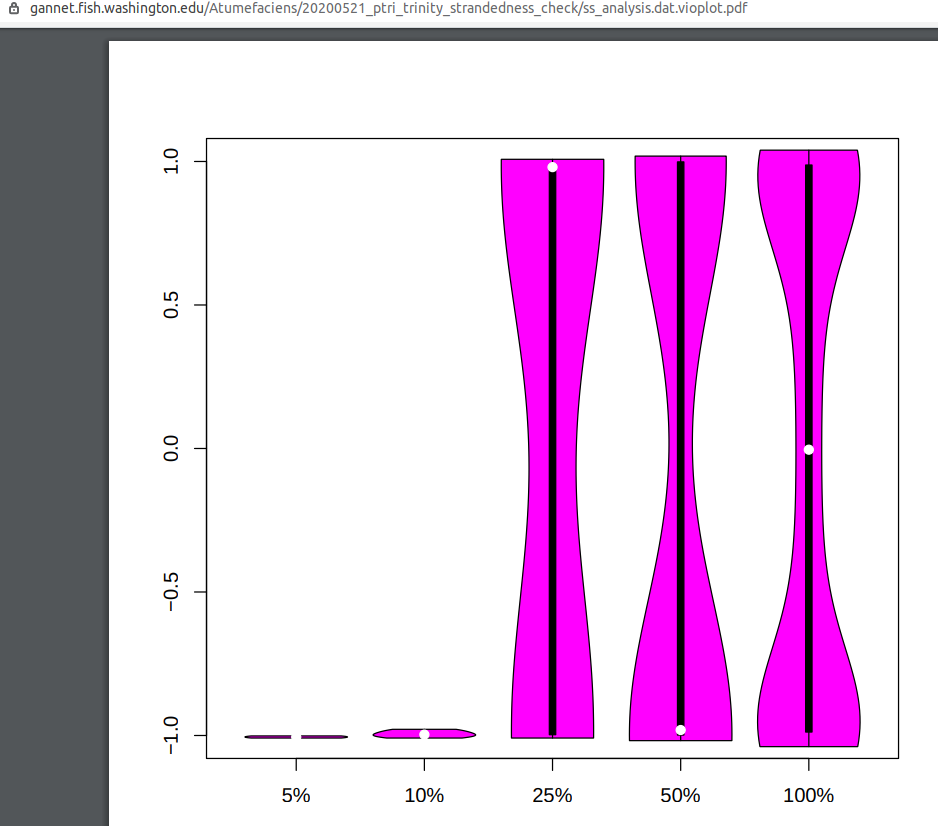
This took a surprisingly long time to run, considering I was only using a single set of paired-end reads; ~15hrs (note: failure shown below was due to a missin R package (vioplot) needed for plotting)
Output folder:
Violin plots (PDF):
Trinity Example:
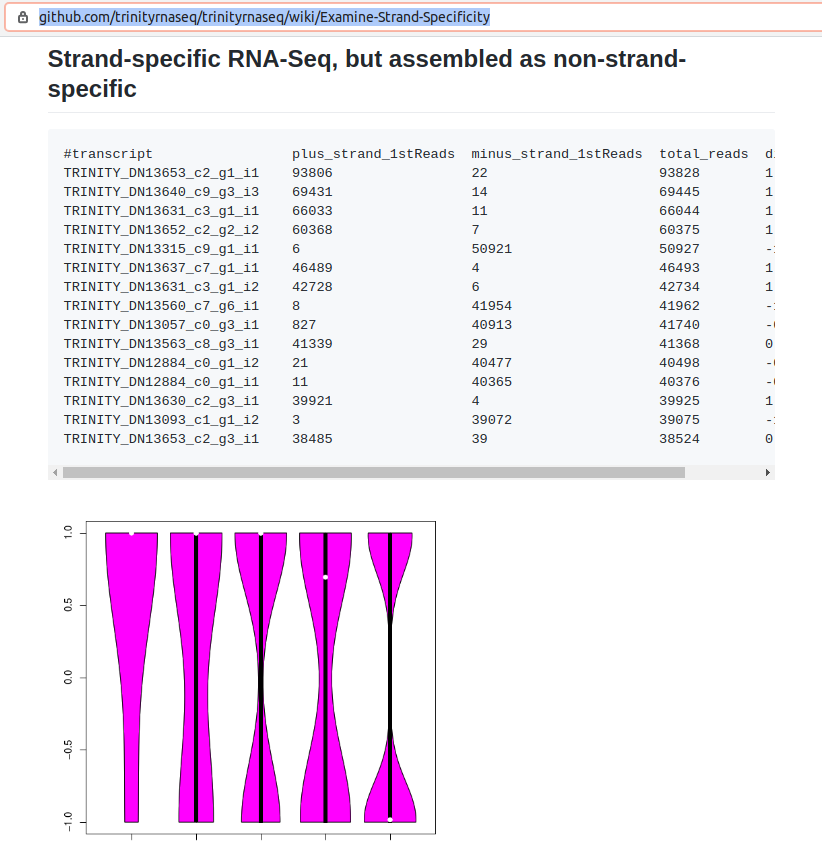
The shape of my violin plots (“barbell”) match the Trinity example of stranded libraries aligned to an assembly created with the non-stranded setting, indicating that the libraries for these RNAseq reads are stranded. I will create a transcriptome assembly from all of the reads using the stranded setting in Trinity.