I was looking for some crab transcriptomic data today and, unable to find any previously assembled transcriptomes, turned to the good ol’ NCBI SRA. In order to simplify retrieval and conversion of SRA data, need to use the SRA Toolkit software suite. Since I haven’t used this in many years, I figured I might as well put together a brief guide/tutorial so I can refer back to it in the future.
It should be noted that this is written to describe usage of the SRA Toolkit on our Mox account (UW HPC). If setting this up elsewhere, you’ll want (need?) to configure the default storage location that the SRA Toolkit will use on your specific computer.
As a side note, I found this helpful page which tracks arthropod genome data present on NCBI:
Start by visiting the SRA BioProject page for a particular SRA.
- BioProject Page:

- Click on the “Number of Links” in “SRA Experiments” row:
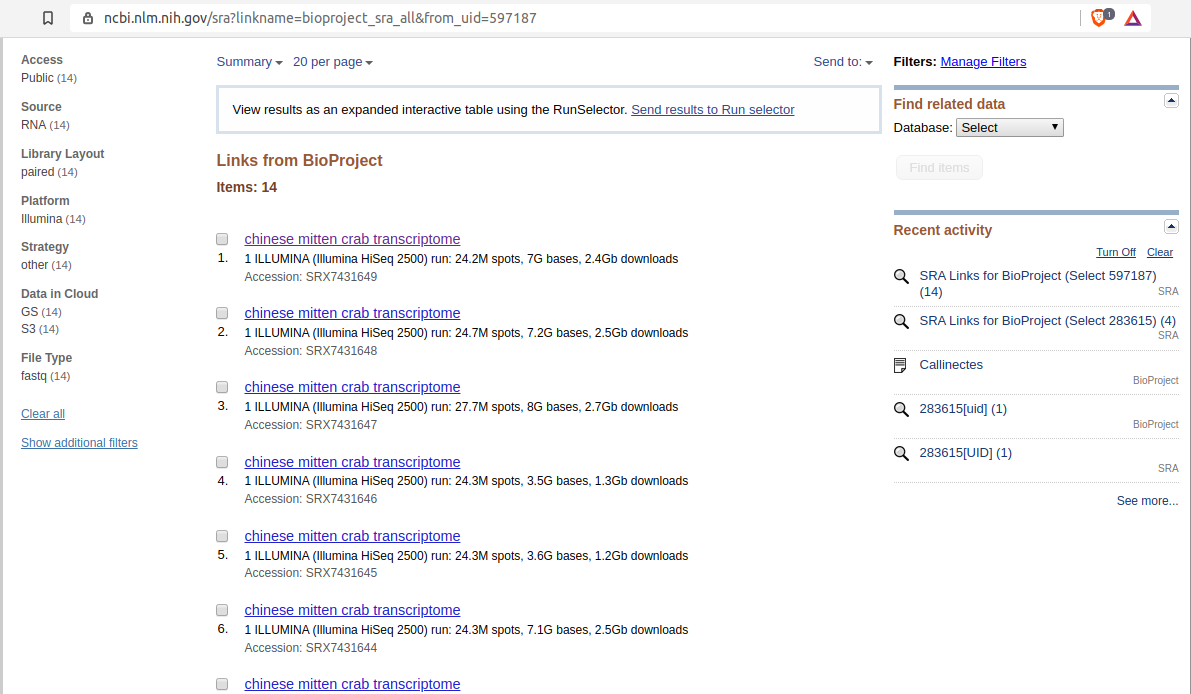
- Click on “All runs” link:
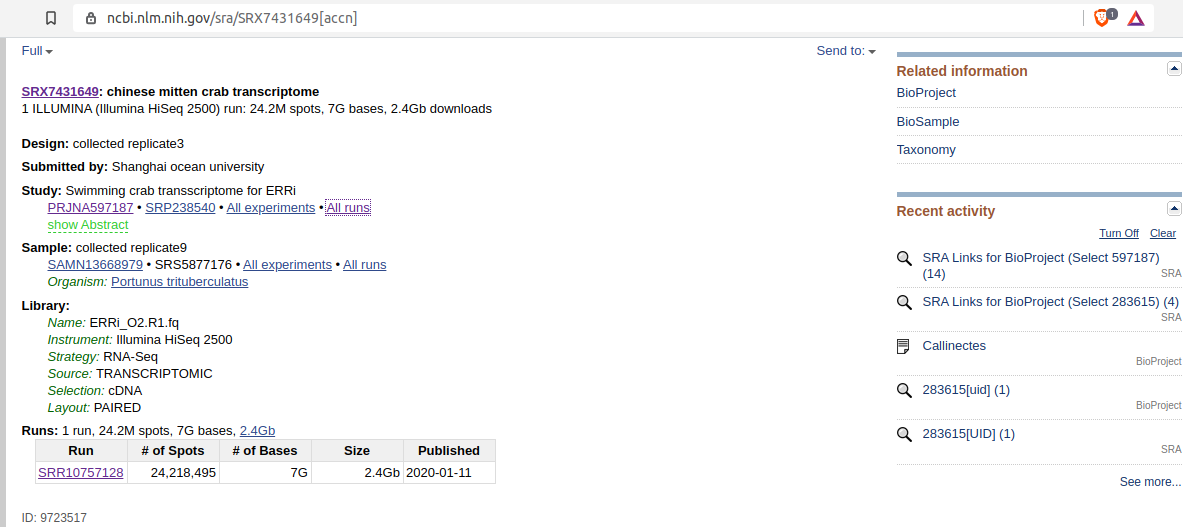
- Click on “Accesion List” (circled) to download text file of all associated SRR accessions:

That file will look like this:
$ head SRR_Acc_List.txt
SRR10757136
SRR10757128
SRR10757129
SRR10757130
SRR10757131
SRR10757132
SRR10757133
SRR10757134
SRR10757135
SRR10757137Use that file to download the actual SRA files.
/gscratch/srlab/programs/sratoolkit.2.10.6-centos_linux64/bin/prefetch.2.10.6 --output-directory . --option-file SRR_Acc_List.txtIf running on Mox, you’ll need to use a build node, as the processing will be more than allowed with the default login node and you need internet access (which is not avaiable on an interactive/execute node).
If no output directory is specified, the files will end up in:
/gscratch/srlab/data/ncbi/sra/
Get FastQ files from the SRA file(s). This can be run on a build node, an interactive node, or via an execute node using an SBATCH script. The settings used in the example below will produce a set of paired FastQ files for each SRA file (assuming the SRA consists of paired-end reads).
for file in *.sra
do
/gscratch/srlab/programs/sratoolkit.2.10.6-centos_linux64/bin/fasterq-dump.2.10.6 \
--outdir . \
--split-files \
--threads 27 \
--mem 100GB \
--progres \
${file}
done