Mac was getting some weird results when mapping some single cell RNAseq data to the C.gigas mitochondrial (mt) genome that she had, so she asked for some help mapping other C.gigas RNAseq data (GitHub Issue) to the C.gigas mt genome to see if someone else would get similar results.
Per Mac’s suggestion, I used STAR to perform an RNAseq alignment.
I used a genome FastA and transcriptome GTF file that she had previously provided in this GitHub Issue, so I don’t know much about their origination/history.
For RNAseq data, I used the only Roberts Lab C.gigas data I could find (see Nightingales (Google Sheet) for more info), which was surprisingly limited. I didn’t realize that we’ve performed so few RNAseq experiments with C.gigas.
I used the following files for the alignment:
RNAseq (FastQ):
2M_AGTCAA_L001_R1_001.fastq.gz (2.4GB)
4M_AGTTCC_L001_R1_001.fastq.gz (2.0GB)
http://owl.fish.washington.edu/nightingales/C_gigas/4M-HS_GTCCGC_L001_R1_001.fastq.gz (1.5GB)
http://owl.fish.washington.edu/nightingales/C_gigas/6M_ATGTCA_L001_R1_001.fastq.gz (2.0GB)
http://owl.fish.washington.edu/nightingales/C_gigas/6M-HS_GTGAAA_L001_R1_001.fastq.gz (1.5GB)
Genome FastA (540MB):
Transcriptome GTF (380MB):
This was run on Mox.
SBATCH script (GitHub):
#!/bin/bash
## Job Name
#SBATCH --job-name=20201208_cgig_STAR_RNAseq-to-NCBI-GCF_000297895.1_oyster_v9
## Allocation Definition
#SBATCH --account=coenv
#SBATCH --partition=coenv
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=10-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20201208_cgig_STAR_RNAseq-to-NCBI-GCF_000297895.1_oyster_v9
### C.gigas RNAseq alignment to NCBI genome FastA file from Mac GCF_000297895.1_oyster_v9_genomic.fasta.
### Mackenzie Gavery asked for help to evaluate RNAseq read mappings to mt genome.
###################################################################################
# These variables need to be set by user
# Working directory
wd=$(pwd)
# Set number of CPUs to use
threads=28
# Initialize arrays
fastq_array=()
# Input/output files
fastq_checksums=fastq_checksums.md5
genome_fasta_checksum=genome_fasta_checksum.md5
gtf_checksum=gtf_checksum.md5
rnaseq_reads_dir=/gscratch/srlab/sam/data/C_gigas/RNAseq
gtf=/gscratch/srlab/sam/data/C_gigas/transcriptomes/GCF_000297895.1_oyster_v9_genomic.gtf.wl_keep_mito_v7.sorted.gtf
genome_dir=${wd}/genome_dir
genome_fasta=/gscratch/srlab/sam/data/C_gigas/genomes/GCF_000297895.1_oyster_v9_genomic.fasta
# Paths to programs
multiqc=/gscratch/srlab/programs/anaconda3/bin/multiqc
samtools="/gscratch/srlab/programs/samtools-1.10/samtools"
star=/gscratch/srlab/programs/STAR-2.7.6a/bin/Linux_x86_64_static/STAR
# Programs associative array
declare -A programs_array
programs_array=(
[multiqc]="${multiqc}" \
[samtools_index]="${samtools} index" \
[samtools_sort]="${samtools} sort" \
[samtools_view]="${samtools} view" \
[star]="${star}"
)
###################################################################################
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Load GCC OMP compiler. Might/not be needed for STAR
module load gcc_8.2.1-ompi_4.0.2
# Make STAR genome directory
mkdir --parents ${genome_dir}
# Populate RNAseq array
fastq_array=(${rnaseq_reads_dir}/*.fastq)
# Comma separated list required for STAR mapping
# Uses tr to change spaces between elements to commas
fastq_list=$(tr ' ' ',' <<< "${fastq_array[@]}")
# Create STAR genome indexes
# Overhang value is set to "generic" 100bp -
# this value is unknown and is the suggested default in
# STAR documentation.
${programs_array[star]} \
--runThreadN ${threads} \
--runMode genomeGenerate \
--genomeDir ${genome_dir} \
--genomeFastaFiles ${genome_fasta} \
--sjdbGTFfile ${gtf} \
--sjdbOverhang 100 \
--genomeSAindexNbases 13
# Run STAR mapping
# Sets output to sorted BAM file
${programs_array[star]} \
--runThreadN ${threads} \
--genomeDir ${genome_dir} \
--outSAMtype BAM SortedByCoordinate \
--readFilesIn ${fastq_list}
# Index BAM output file
${programs_array[samtools_index]} \
Aligned.sortedByCoord.out.bam
# Extract mt alignments
# -h: includes header
${programs_array[samtools_view]} \
--threads ${threads} \
--write-index \
-h \
Aligned.sortedByCoord.out.bam NC_001276.1 \
-o Aligned.sortedByCoord.out.NC_001276.1.bam
# Generate checksums for reference
# Uses bash string substitution to replace commas with spaces
# NOTE: do NOT quote string substitution command
for fastq in ${fastq_list//,/ }
do
# Generate MD5 checksums for each input FastQ file
echo "Generating MD5 checksum for ${fastq}."
md5sum "${fastq}" >> "${fastq_checksums}"
echo "Completed: MD5 checksum for ${fastq}."
echo ""
done
# Run MultiQC
${programs_array[multiqc]} .
# Generate checksums for genome FastA and GTF
echo "Generating MD5 checksum for ${genome_fasta}."
md5sum "${genome_fasta}" > "${genome_fasta_checksum}"
echo "Completed: MD5 checksum for ${genome_fasta}."
echo ""
echo "Generating MD5 hecksum for ${gtf}."
md5sum "${gtf}" > "${gtf_checksum}"
echo "Completed: MD5 checksum for ${gtf}."
echo ""
# Capture program options
echo "Logging program options..."
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${program}: "
echo ""
# Handle samtools help menus
if [[ "${program}" == "samtools_index" ]] \
|| [[ "${program}" == "samtools_sort" ]] \
|| [[ "${program}" == "samtools_view" ]]
then
${programs_array[$program]}
fi
${programs_array[$program]} -h
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
# If MultiQC is in programs_array, copy the config file to this directory.
if [[ "${program}" == "multiqc" ]]; then
cp --preserve ~/.multiqc_config.yaml multiqc_config.yaml
fi
done
echo ""
echo "Finished logging program options."
echo ""
echo ""
echo "Logging system PATH."
# Document programs in PATH (primarily for program version ID)
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
echo "Finished logging system PATH"RESULTS
This was pretty quick, but wasn’t really sure what to expect. Only 20mins:

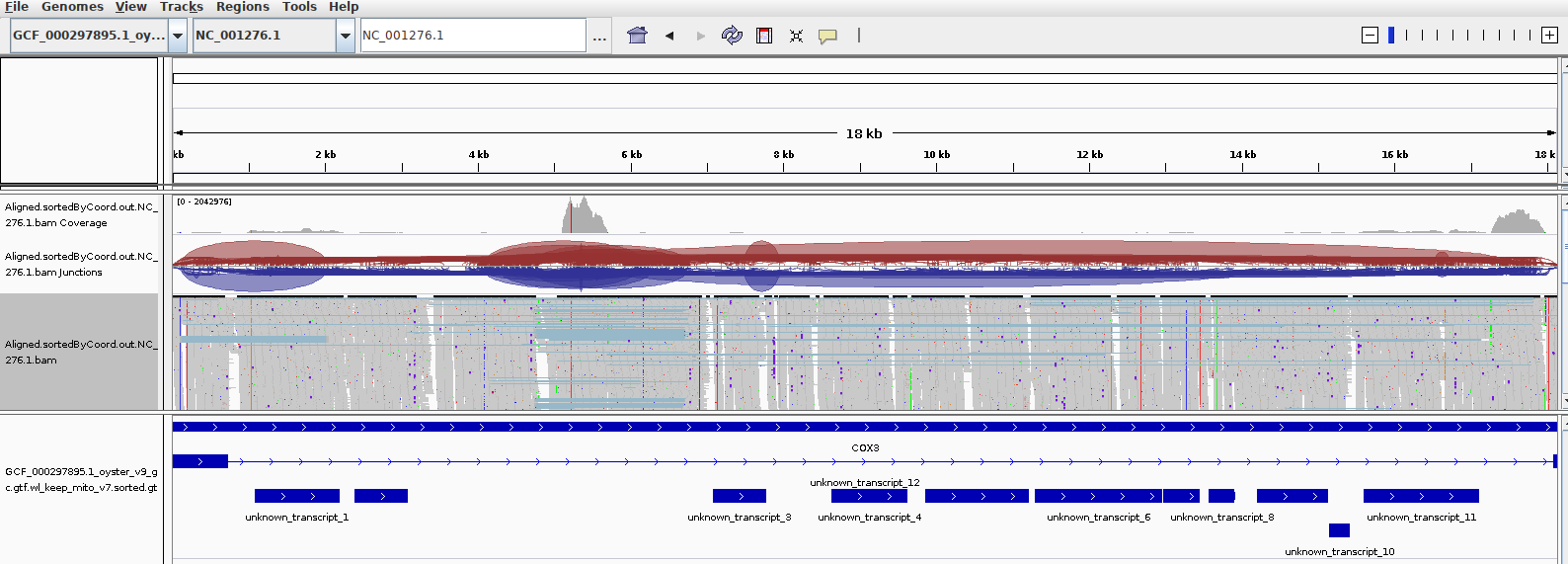
A quick comparison of my alignment with what Mac saw previously show very similar results:
SAM’S ALIGNMENT:
MAC’S ALIGNMENT:

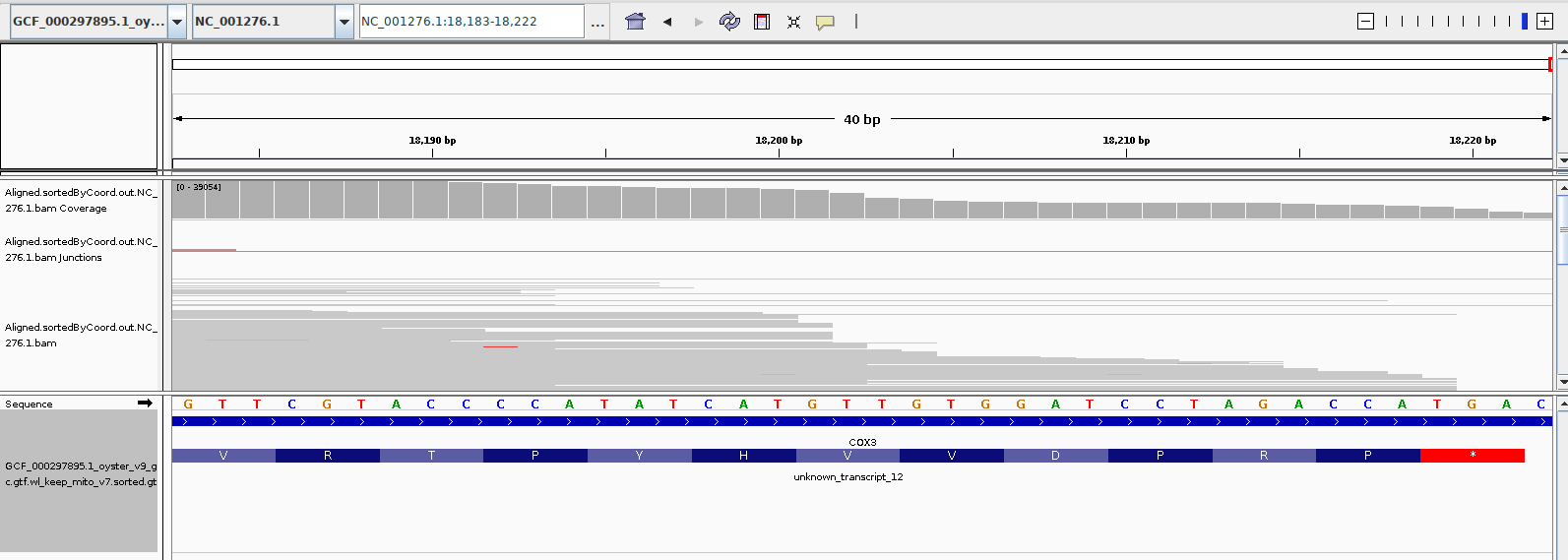
Her primary concern is the fact that a majority of the reads appear to align to non-coding regions of the C.gigas mt genome. My alignment shows the same. I suspect that is likely related to poor annotation of the C.gigas mt genome/transcriptome. Also, I believe the mitochondrial translation codons differ from that of nuclear translation codons. On top of that, I think invertebrates might also have a slightly altered set of translation codons. Zooming in on the IGV alignment seems to show that the standard (mammalian) codons were used to identify coding regions.
Notice that the stop codon from this alignment shown below uses TGA as termination. In invertebrate mt genomes, this codon actually encodes for tryptophan (Trp/W). This suggests that the GTF file was generated with a standard (i.e. vertebrate, non-mitochondrial) codon table, instead of a mt codon table (and almost certainly not an invertebrate mt codon table).
In any case, I’ve posted my thoughts/results in that GitHub Issue. Links to files are below.
Output folder:
20201208_cgig_STAR_RNAseq-to-NCBI-GCF_000297895.1_oyster_v9/
BAM files:
Mitochondrial BAM and index:
Full BAM and index:
MD5 checksums (TEXT):