In preparation for FastQC and trimming of the E5 coral sRNA-seq data, I noticed that my “default” trimming settings didn’t produce the results I expected. Specifically, since these are sRNAs and the NEBNext® Multiplex Small RNA Library Prep Set for Illumina (PDF) protocol indicates that the sRNAs should be ~21 - 30bp, it seemed odd that I was still ending up with read lengths of 150bp. So, I tried a couple of quick trimming comparisons on just a single pair of sRNA FastQs to use as examples to get feeback on how trimming should proceed.
Trimming was done with the flexbar. As an aside, I might begin using this trimmer instead of fastp going forward. fastp has some odd “quirks” in it’s order of operations that sometimes require two rounds of trimming. Also, it’s annoying that fastp limits the number of threads to 16; flexbar has no such limitation. Perhaps this is moot, as I’m not sure if there’s truly a performance increase or not. The biggest trade off, though, is that fastp automatically generates HTML reports for trimming, which include pre- and post-trimming plots/data. These are very useful and are also interpreted by MultiQC…
This was all done on Raven using a Jupyter Notebook.
Jupyter Notebook (GitHub):
Jupyter Notebook (NB Viewer):
RESULTS
Output folder:
20230524-E5-coral-sRNAseq_trimmings_comparisons
MultiQC Report (HTML)
Adapter Trim Only FastQC Reports (HTML)
https://gannet.fish.washington.edu/Atumefaciens/20230524-E5-coral-sRNAseq_trimmings_comparisons/sRNA-ACR-140-S1-TP2_R1_001-adapter_trim_only_1_fastqc.html
https://gannet.fish.washington.edu/Atumefaciens/20230524-E5-coral-sRNAseq_trimmings_comparisons/sRNA-ACR-140-S1-TP2_R1_001-adapter_trim_only_2_fastqc.html
Adapter and 50bp length trim FastQC Reports (HTML)
https://gannet.fish.washington.edu/Atumefaciens/20230524-E5-coral-sRNAseq_trimmings_comparisons/sRNA-ACR-140-S1-TP2_R1_001-adapter-and-length-50_1_fastqc.html
https://gannet.fish.washington.edu/Atumefaciens/20230524-E5-coral-sRNAseq_trimmings_comparisons/sRNA-ACR-140-S1-TP2_R1_001-adapter-and-length-50_2_fastqc.html
Let’s take a brief look at the data:
Adapter trimming only
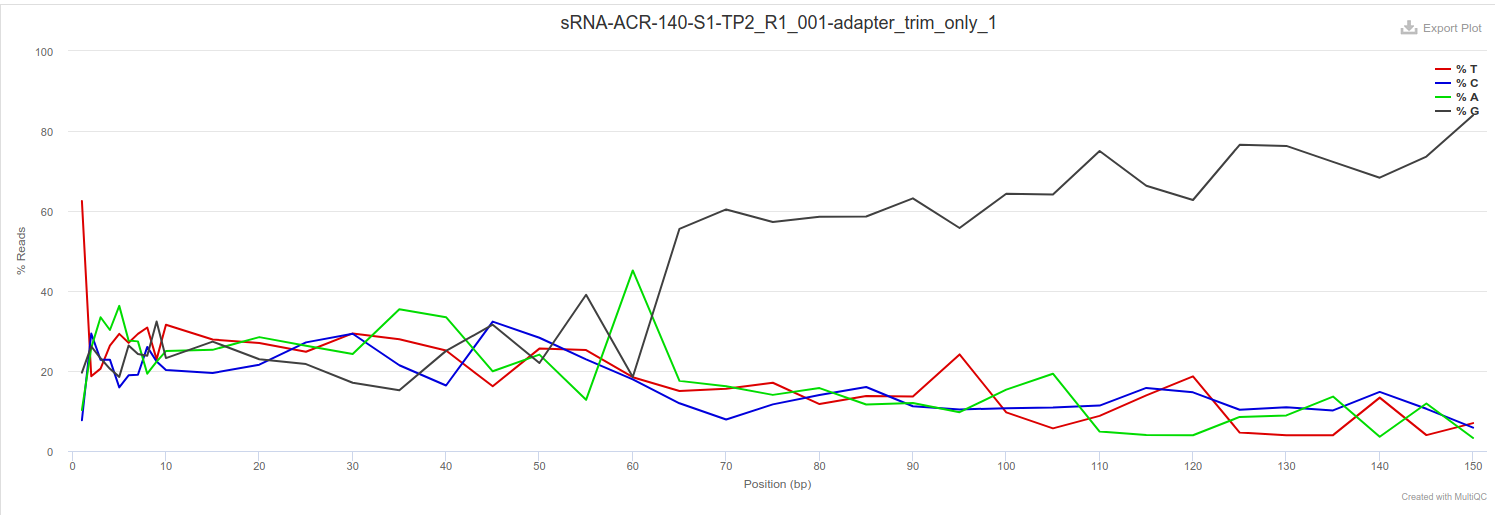
FastQC of adapter trim only still shows read lengths of 150bp. Additionally, the bulk of the 3’ end of the reads show extensive poly-G signals. Admittedly, flexbar doesn’t have a default poly-G trimming option. However, using fastp, which does have a poly-G trimming option, still showed similar results (data not shown - not comparing trimmers, just highlighting persistence of long reads).
Adapter and length trimming
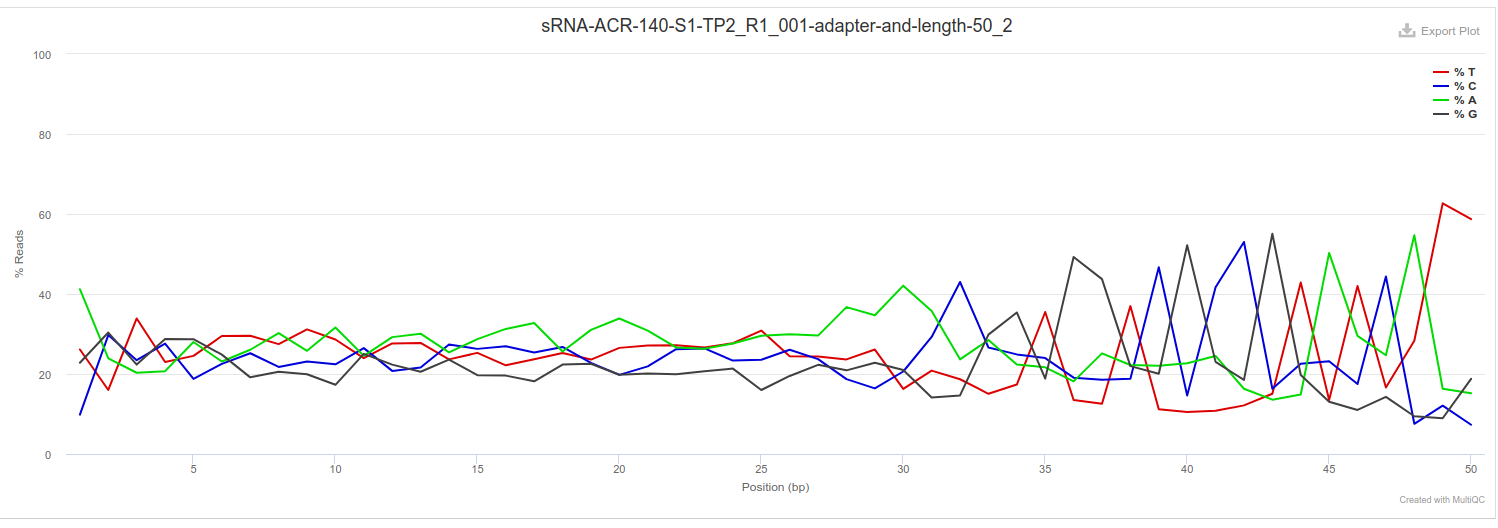
FastQC of adapter trim and trimming to a length of 50bp (from the 3’ end). As expected, performing length trimming removed all reads longer than 50bp, which also resulted in removal of poly-G sequence. Also shows an increase in heterogeneity (i.e. more drastic spikes in plots) after ~30bp. This is probably expected, as the NEBNext® Multiplex Small RNA Library Prep Set for Illumina (PDF) manual indicates that miRNA should be ~21bp and piRNAs ~31bp. Thus, the sequence after that could be something else.
Will share with E5 group to get feedback.