Intro
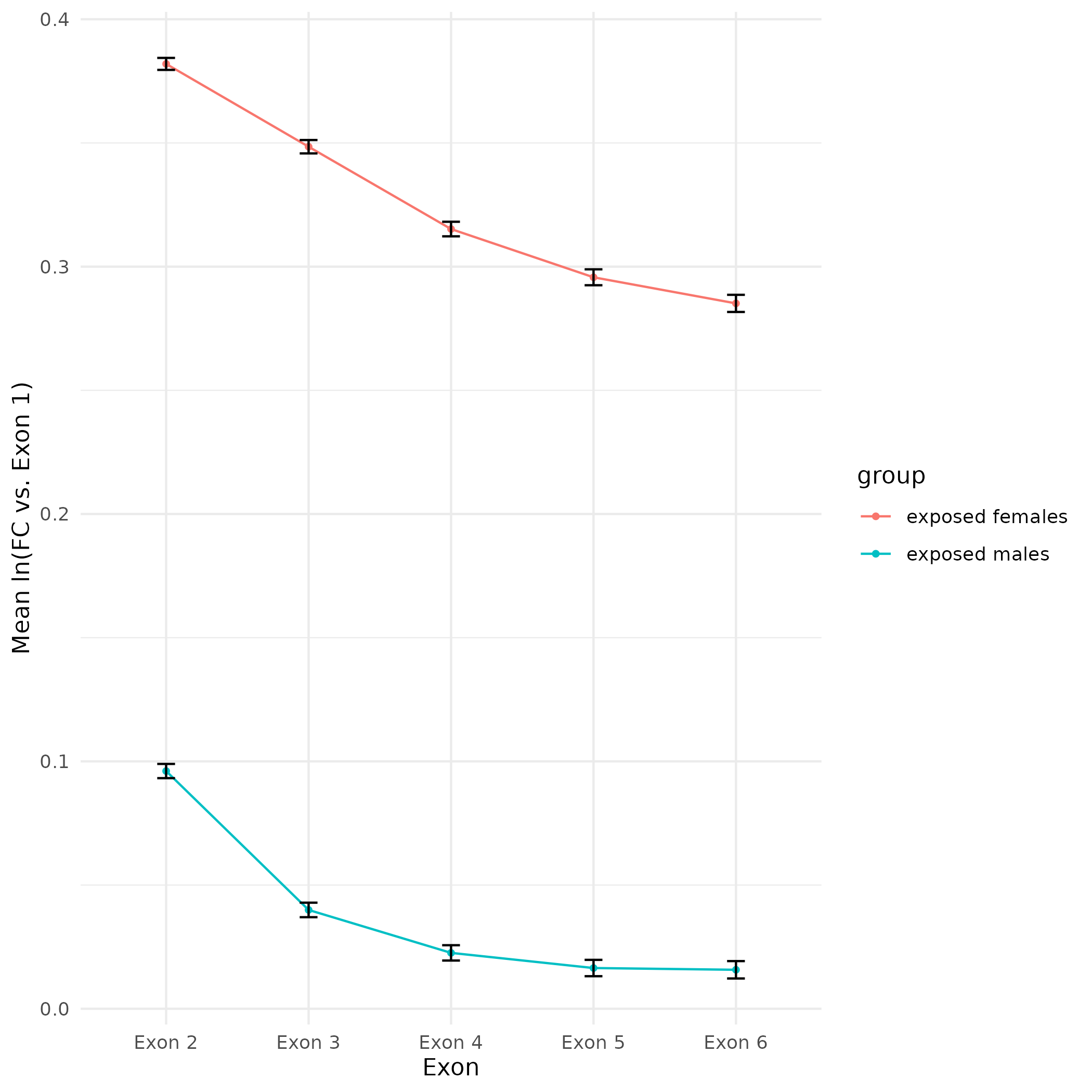
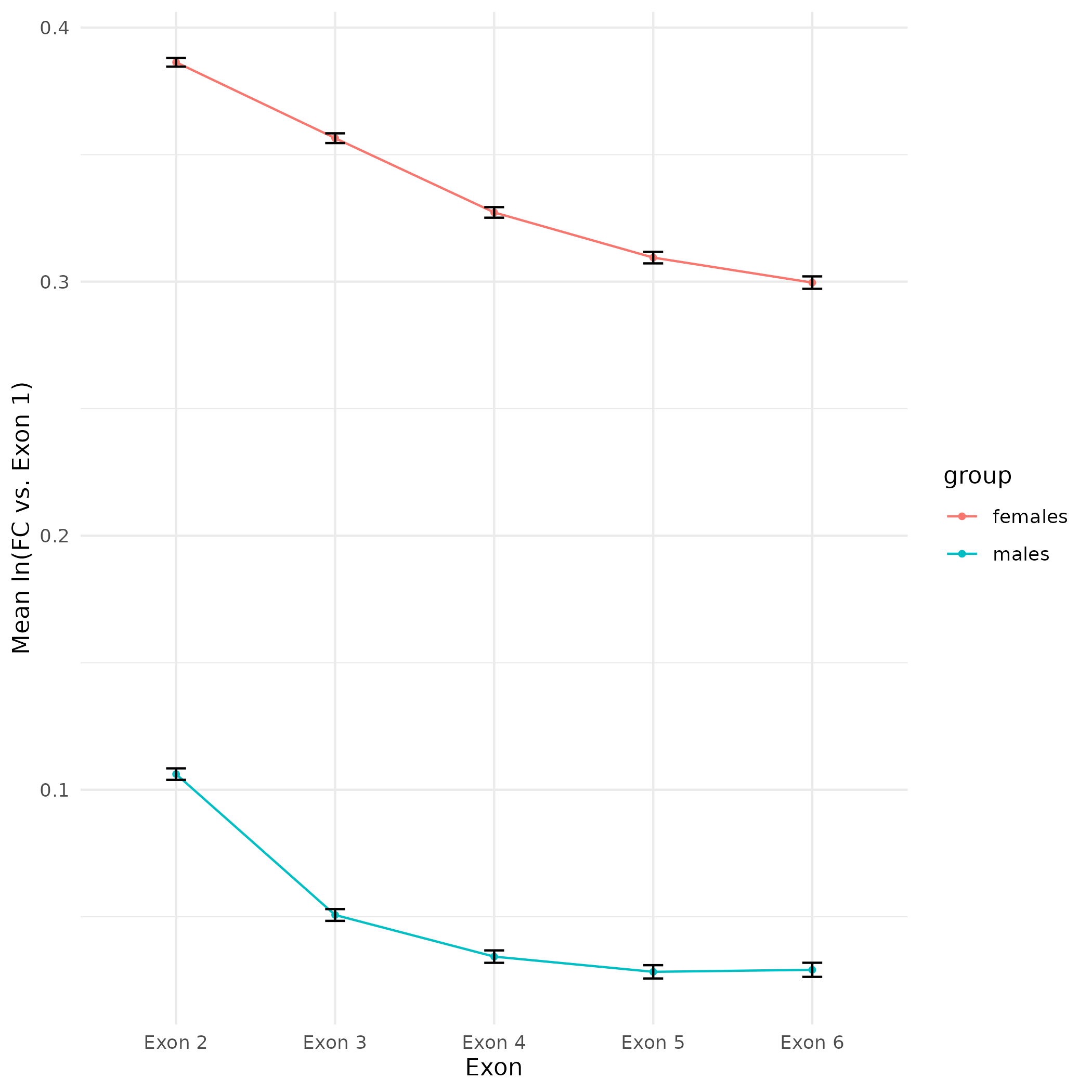
As part of the CEABIGR project (GitHub repo) Steven performed some inital data wrangling to test out the basic calculations to determine the natural log of the fold change in exon expression, relative to Exon 1, for each gene in a single sample. The decision to perform the calculation in this manner was based on (Li et al. 2018). I plotted the results previously (on 20240103), but that did not include any sort of “normalization” across samples. Further discussion (GitHub Issue) decided to set a threshold based on the sum of reads for Exons 1 -6 per gene, per sample. The value decided on was a minimum sum of 10 reads for Exons 1-6, per gene, across all samples. E.g. If S13M has a sum of 15 reads for Exons 1-6 for a given gene, but S9M only has a sum of 8 reads for Exons 1-6 for that same gene, then that gene is discarded from the fold chane calculations.
This threshold filtering retained 23,101 genes (out of 38,264; 60.4% of genes)
After this, will explore how spurious transcription relates to methylation levels across genes, using this new exon sum threshold.
See 65-exon-coverage.qmd for code.
Code and plots link to commit 0b9a89a.
Plots
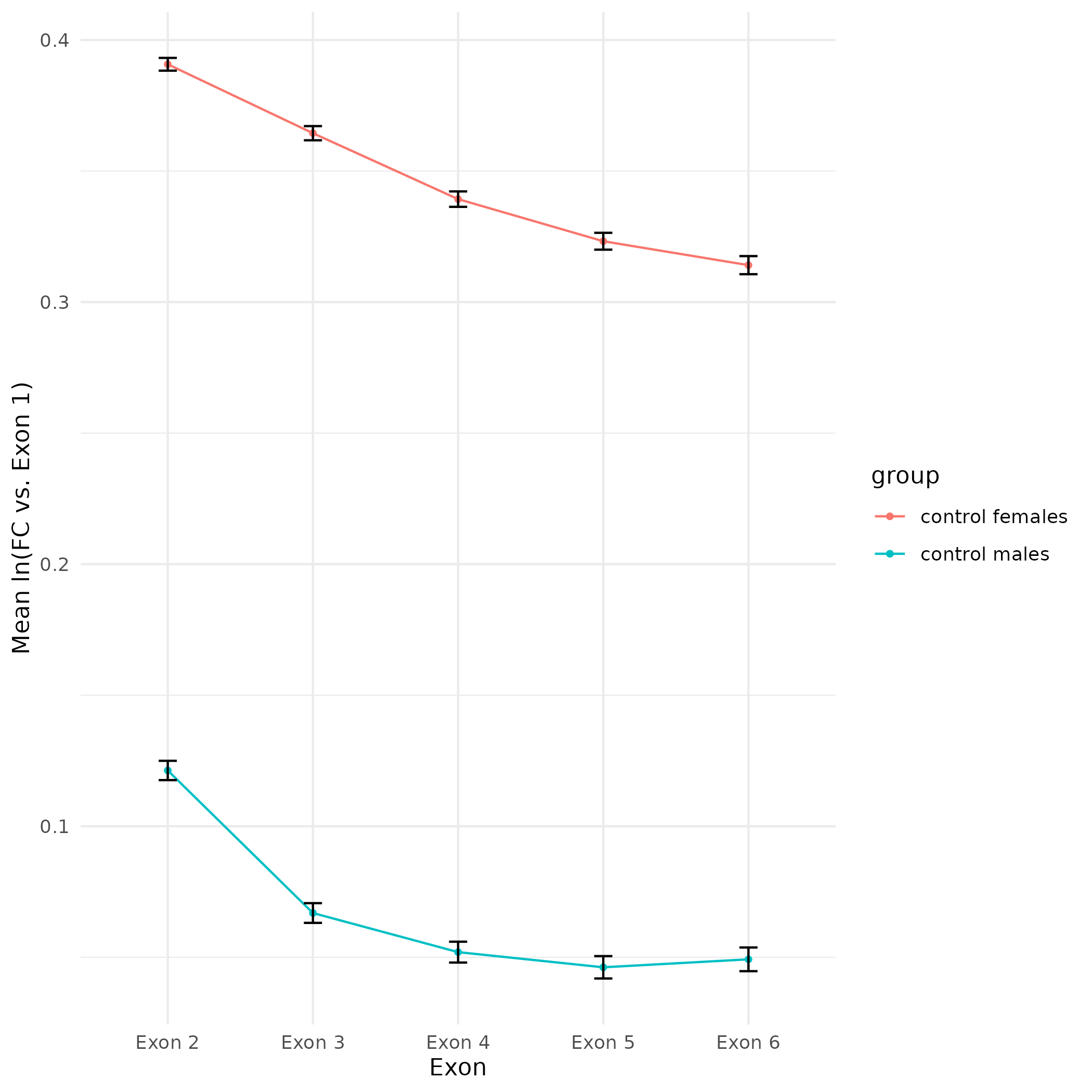
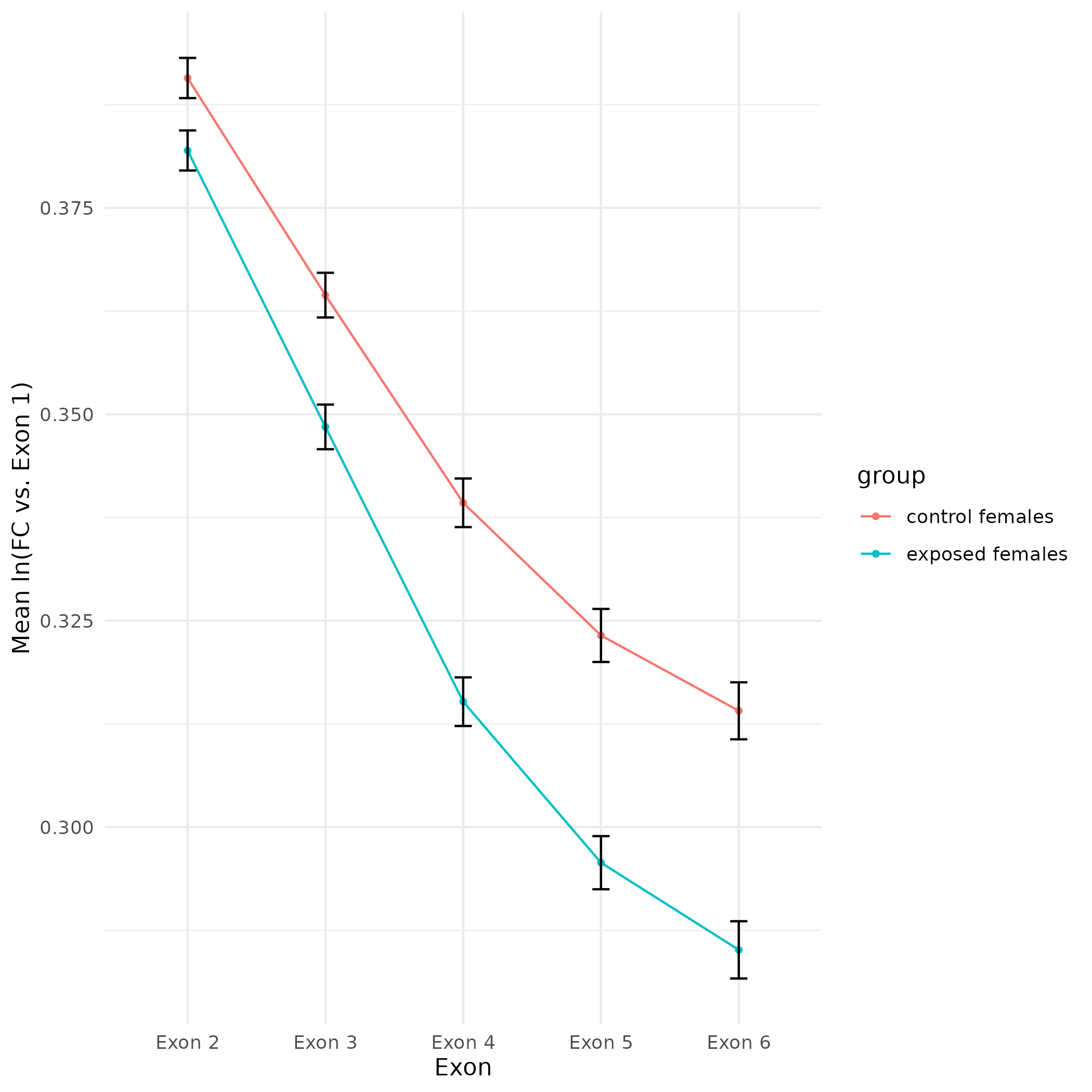
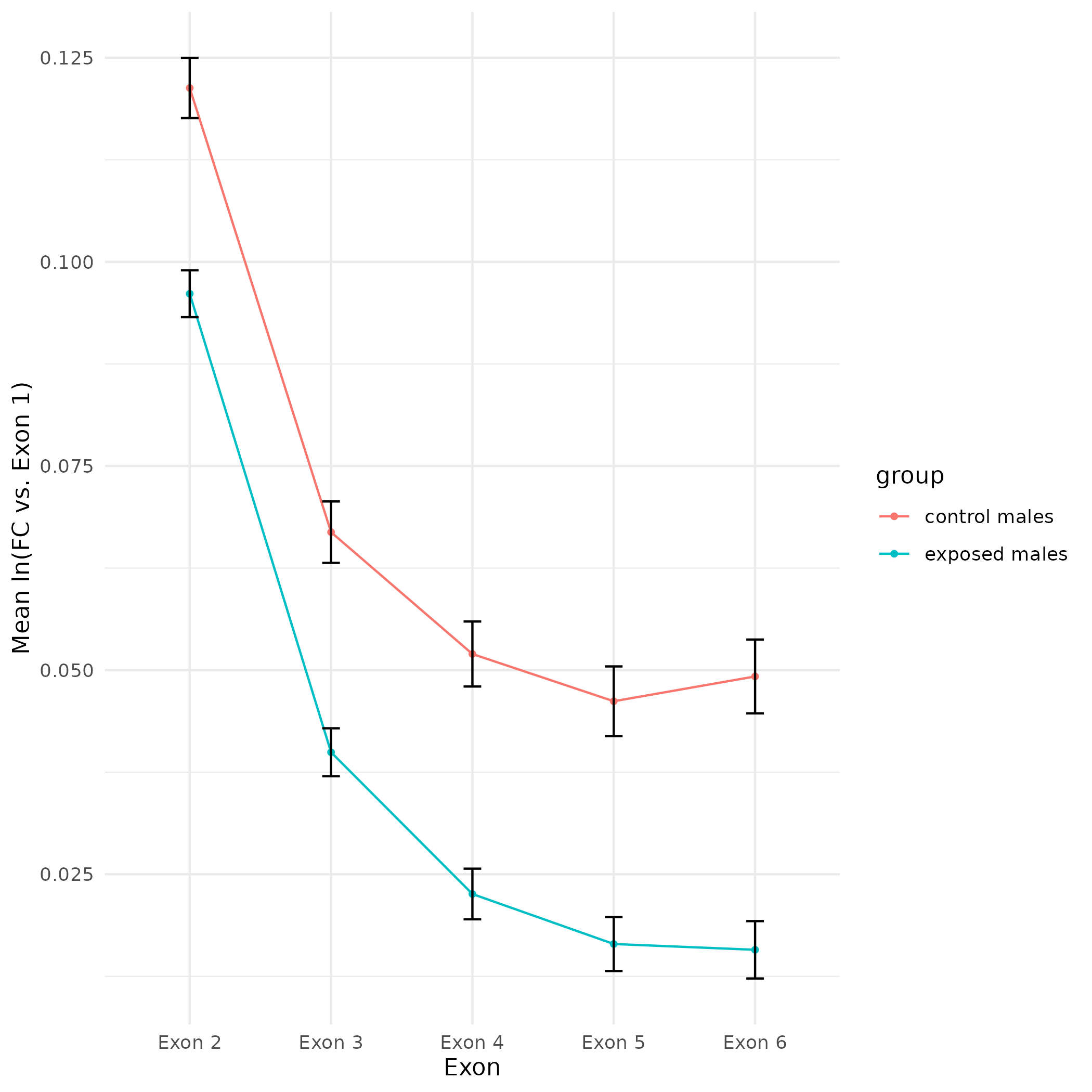
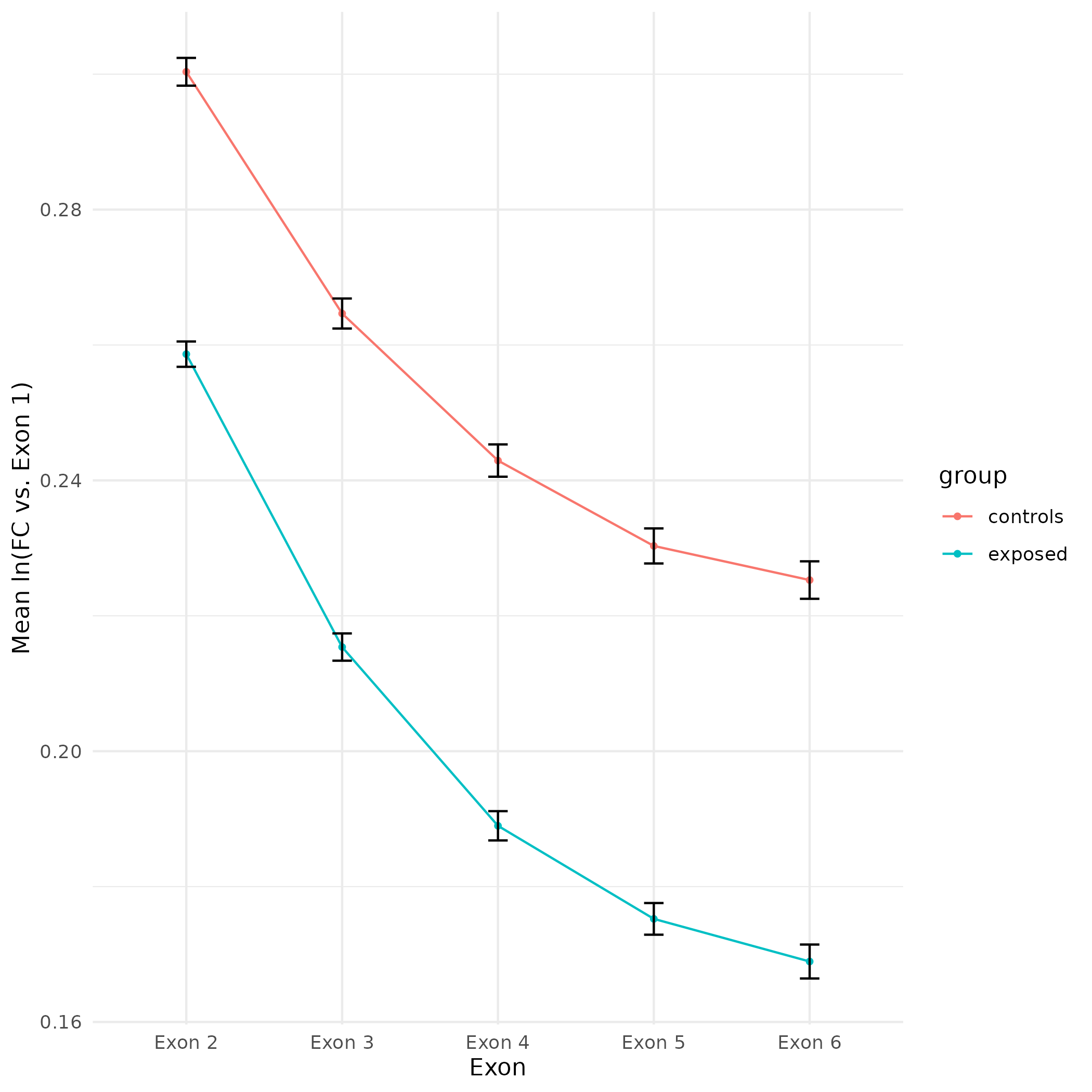
All plots are line plots of the mean natural log fold-change in exon expression (Exons 2-6), relative to Exon 1. All genes used in the analysis had to Black bars represent standard error.
Plots are simply arranged side-by-side. Scales of axes are not intended to match.