INTRO
In preparation for a grant proposal involving sea star wasting disease, due in about two weeks, Steven asked that I perform taxonomic classification on unmapped P.helanthoides RNA-seq reads (GitHub Issue). Due to time constraints, and a bit of luck, I opted to run this on Mox (Mox should have been decommissioned on 11/1/2024, but it’s still running and being able to use Mox, which is already set up, instead of resorting to Klone, was much faster to get this going). Also, due to time constraints, Steven suggested I run this on just a single set of reads (GitHub Issue comment). I opted for the following, since this analysis had already been started and was already working on these:
PSC-0519_unmapped_reads.[12].fastq
METHODS
DIAMOND BLASTx and MEGANization
Reads were DIAMOND BLASTx’d against a previously constructed DIAMON BLAST database. After DIAMOND BLASTx, the resulting DAA files were “MEGANized” in preparation for use in MEGAN6. Both steps were run on Mox.
SBATCH SCRIPT
GitHub:
#!/bin/bash
## Job Name
#SBATCH --job-name=20241105-phel-diamond-meganizer
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Resources
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=7-00:00:00
## Memory per node
#SBATCH --mem=120G
##turn on e-mail notification
#SBATCH --mail-type=ALL
#SBATCH --mail-user=samwhite@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/scrubbed/samwhite/outputs/20241105-phel-diamond-meganizer
## Perform DIAMOND BLASTx on unmapped P.helanthoides RNA-seq files from Steven.
## Will be used to view taxonomic breakdown of sequencing reads.
## Expects input FastQ files to be match this pattern: *reads.[12].fastq
###################################################################################
# These variables need to be set by user
## Assign Variables
fastq_pattern='*reads.[12].fastq'
# Program paths
diamond=/gscratch/srlab/programs/diamond-v2.1.1/diamond
meganizer=/gscratch/srlab/programs/MEGAN-6.22.0/tools/daa-meganizer
# DIAMOND NCBI nr database
dmnd_db=/gscratch/srlab/blastdbs/20230215-ncbi-nr/20230215-ncbi-nr.dmnd
# MEGAN mapping files
megan_mapping_dir=/gscratch/srlab/sam/data/databases/MEGAN
megan_mapdb="${megan_mapping_dir}/megan-map-Feb2022.db"
# FastQ files directory
fastq_dir=/gscratch/scrubbed/samwhite/data/P_helianthoides/RNAseq
# CPU threads
threads=40
# MEGAN memory limit
mem_limit=100G
# Programs associative array
declare -A programs_array
programs_array=(
[diamond]="${diamond}" \
[meganizer]="${meganizer}"
)
###################################################################################################
# Exit script if any command fails
set -e
# Load Python Mox module for Python module availability
module load intel-python3_2017
# Loop through FastQ files, log filenames to fastq_list.txt.
# Run DIAMOND on each FastQ, followed by "MEGANization"
# DO NOT QUOTE ${fastq_pattern}
for fastq in "${fastq_dir}"/${fastq_pattern}
do
# Log input FastQs
echo ""
echo "Generating MD5 checksum for ${fastq}..."
md5sum "${fastq}" | tee --append input_fastqs-checksums.md5
echo ""
# Strip leading path ${fastq##*/} by eliminating all text up to and including last slash from the left side.
# Strip extensions by eliminating ".fastq.gz" from the right side.
no_path=$(echo "${fastq##*/}")
no_ext=$(echo "${no_path%%.fastq}")
# Run DIAMOND with blastx
# Output format 100 produces a DAA binary file for use with MEGAN
echo "Running DIAMOND BLASTx on ${fastq}."
echo ""
"${programs_array[diamond]}" blastx \
--db ${dmnd_db} \
--query "${fastq}" \
--out "${no_ext}".blastx.meganized.daa \
--outfmt 100 \
--top 5 \
--block-size 15.0 \
--index-chunks 4 \
--memory-limit ${mem_limit} \
--threads ${threads}
echo "DIAMOND BLASTx on ${fastq} complete: ${no_ext}.blastx.meganized.daa"
echo ""
# Meganize DAA files
# Used for ability to import into MEGAN6
echo "Now MEGANizing ${no_ext}.blastx.meganized.daa"
"${programs_array[meganizer]}" \
--in "${no_ext}".blastx.meganized.daa \
--threads ${threads} \
--mapDB ${megan_mapdb}
echo "MEGANization of ${no_ext}.blastx.meganized.daa completed."
echo ""
done
# Generate MD5 checksums
for file in *
do
echo ""
echo "Generating MD5 checksums for ${file}:"
md5sum "${file}" | tee --append checksums.md5
echo ""
done
# Generate checksum for MEGAN database(s)
{
md5sum "${megan_mapdb}"
md5sum "${dmnd_db}"
} >> checksums.md5
#######################################################################################################
# Capture program options
if [[ "${#programs_array[@]}" -gt 0 ]]; then
echo "Logging program options..."
for program in "${!programs_array[@]}"
do
{
echo "Program options for ${program}: "
echo ""
# Handle samtools help menus
if [[ "${program}" == "samtools_index" ]] \
|| [[ "${program}" == "samtools_sort" ]] \
|| [[ "${program}" == "samtools_view" ]]
then
${programs_array[$program]}
# Handle DIAMOND BLAST menu
elif [[ "${program}" == "diamond" ]]; then
${programs_array[$program]} help
# Handle NCBI BLASTx menu
elif [[ "${program}" == "blastx" ]]; then
${programs_array[$program]} -help
fi
${programs_array[$program]} -h
echo ""
echo ""
echo "----------------------------------------------"
echo ""
echo ""
} &>> program_options.log || true
# If MultiQC is in programs_array, copy the config file to this directory.
if [[ "${program}" == "multiqc" ]]; then
cp --preserve ~/.multiqc_config.yaml multiqc_config.yaml
fi
done
echo "Finished logging programs options."
echo ""
fi
# Document programs in PATH (primarily for program version ID)
echo "Logging system $PATH..."
{
date
echo ""
echo "System PATH for $SLURM_JOB_ID"
echo ""
printf "%0.s-" {1..10}
echo "${PATH}" | tr : \\n
} >> system_path.log
echo "Finished logging system $PATH."DAA to RMA Conversion
Resulting DAA files were converted to RMA6 format for import and analysis in MEGAN6. Due to time constraints, this was performed on Raven without a script. Command used:
/home/shared/megan-6.24.20/tools/daa2rma \
--in ./PSC-0519_unmapped_reads.1.blastx.meganized.daa ./PSC-0519_unmapped_reads.2.blastx.meganized.daa \
--mapDB ~/data/databases/MEGAN/megan-map-Feb2022.db \
--out ./PSC-0519_unmapped_reads.1.blastx.meganized.rma6 ./PSC-0519_unmapped_reads.2.blastx.meganized.rma6 \
--threads 40 2>&1 | tee --append daa2rma_log.txtRESULTS
The Mox run took just under 16hrs to complete:

DISCUSSION
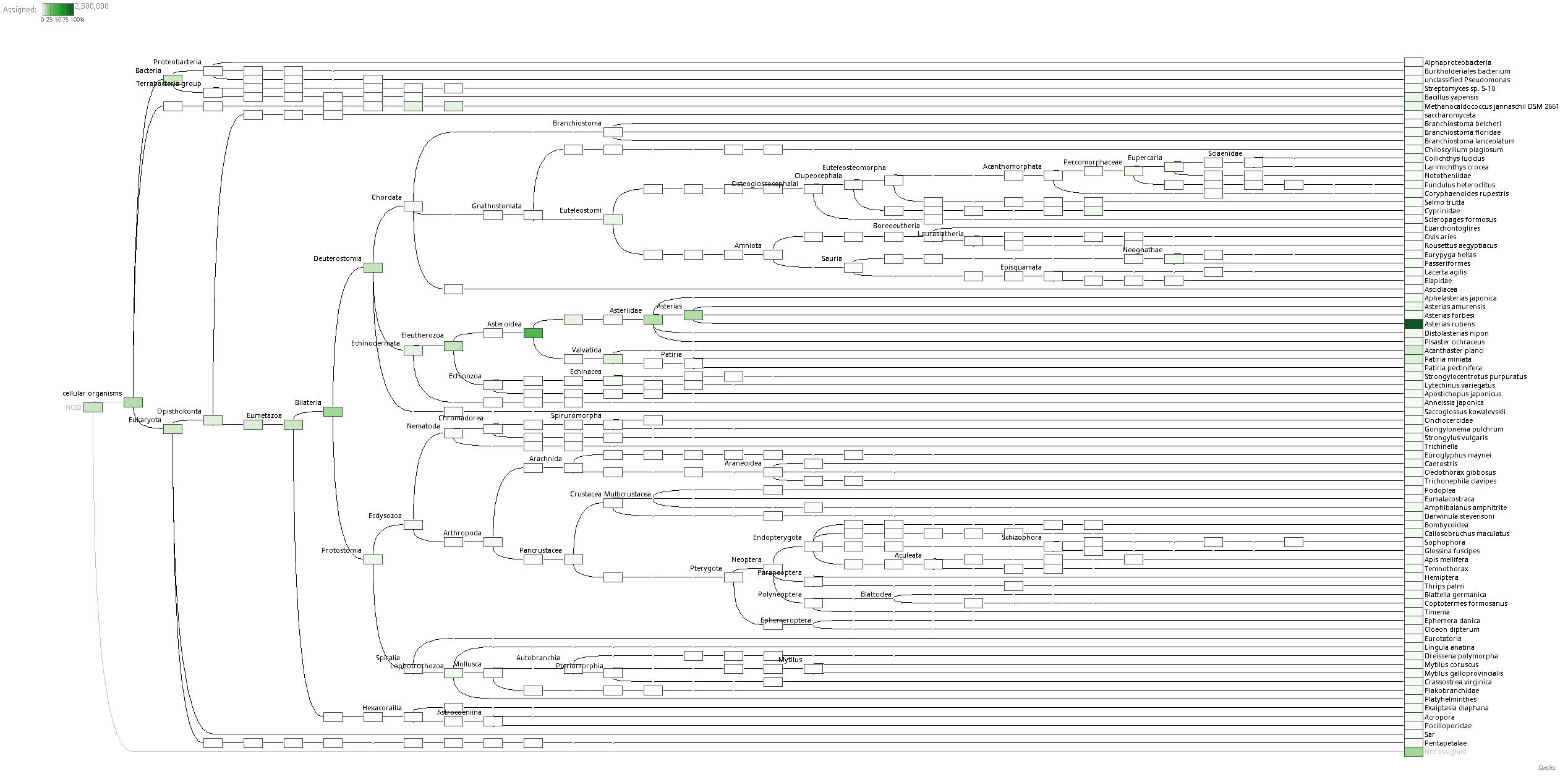
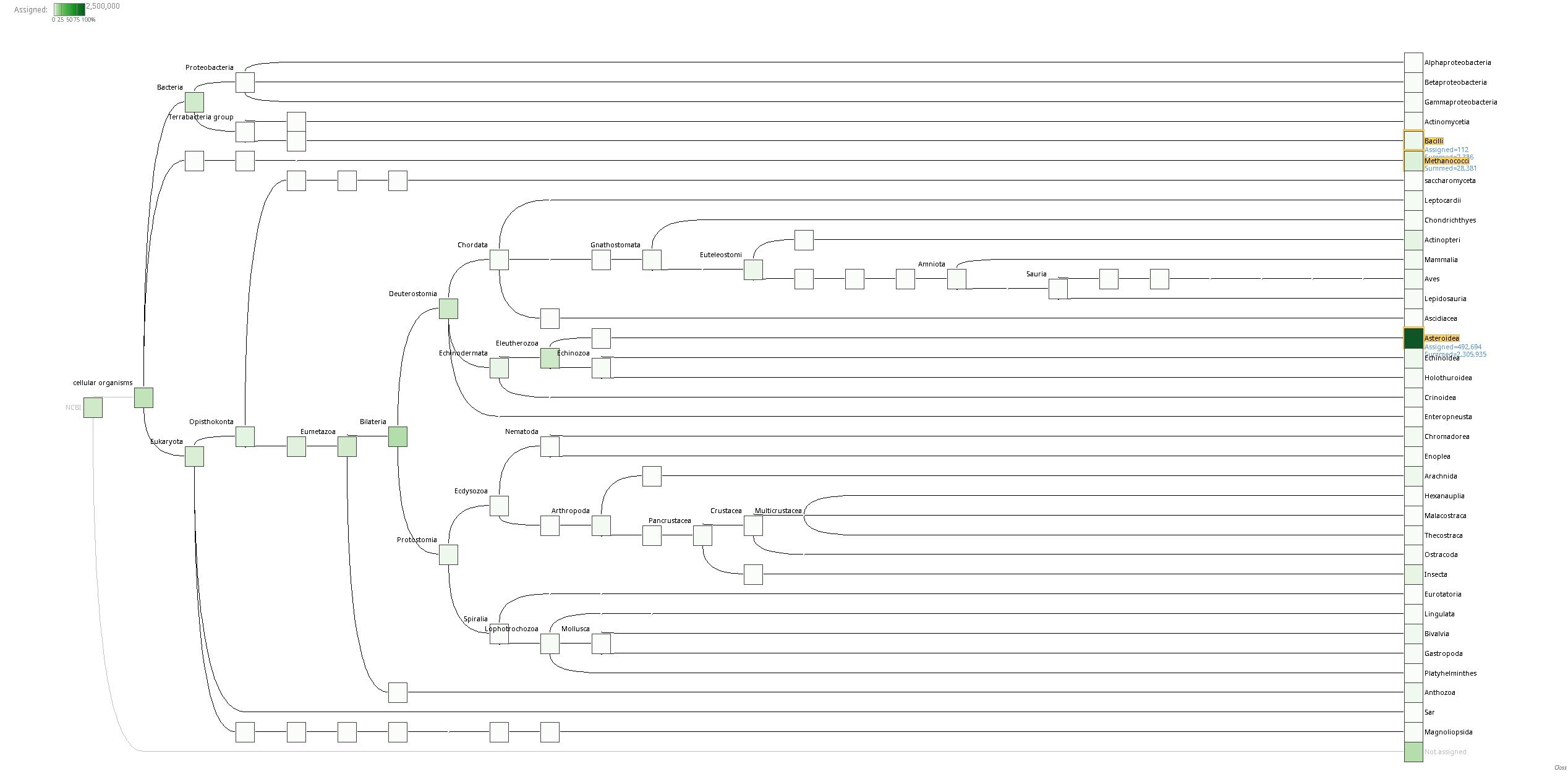
Looking at the entire taxonomies, down to the species level, most of the reads (>1,500,000) get assigned to Asterias rubens (Wikipedia), the common sea star. This is surprising, as I’m fairly certain these sequencing reads came from Pycnopodia helianthoides (Wikipedia), the sunflower star. Of course, these results are also dependent upon how much sequence data in NCBI for any given species. However, with that said, reads are only assigned to a given taxonomic level based on the BLASTx results. Thus, even if a read comes from a sea star, it would (should?) not get assigned to a particular species unless the BLASTx confidence was very high…
Looking at the highest read assignments for non-eukaryotic species, we see that of the Bacteria (7,153 reads), Bacillus yapensis (Journal article website), while in Archae (17,742 reads), Methanocaldococcus jannaschii (Wikipedia) had the most reads assigned.
Output files
Output directory:
DIAMOND BLASTX/MEGANIZER
Output files:
PSC-0519_unmapped_reads.1.blastx.meganized.daa(186GB)- MD5:
1e7207fa8dc3ff15373f46c4e0c5bbb3
- MD5:
PSC-0519_unmapped_reads.2.blastx.meganized.daa(179GB)- MD5:
4e5514926056f0f3273b44da6c3ed7df
- MD5:
RMA6
PSC-0519_unmapped_reads.1.blastx.meganized.rma6- MD5:
b5f0118056d1b3481b5533e420146728
- MD5:
PSC-0519_unmapped_reads.2.blastx.meganized.rma6- MD5:
4e88764010274baccafffe58f67134d2
- MD5: