INTRO
I previously ran FastQC and MultiQC quality checks (20250210) (Notebook) on the P.evermanni raw whole genome bisulfite sequencing (WGBS) data received 20241230 (Notebook), as part of urol-e5/timeseries_molecular (GitHub repo).
These are directional libraries - important info for downstream analysis.
The contents below are from markdown knitted from 01.00-F-Ptua-WGBS-trimming-fastp-FastQC-MultiQC.Rmd (commit 960d871).
1 Background
This notebook will trim raw WGBS FastQs using fastp (Chen 2023). Samples which generate an error during trimming will attempt to be repaired using BBTools repairl.sh (BBMap – Bushnell B. – sourceforge.net/projects/bbmap/). Trimming results will be analyzed with FastQC and summarized by MultiQC (Ewels et al. 2016).
If you need to download the raw sequencing reads, please see 00.20-F-Ptua-WGBS-reads-FastQC-MultiQC.Rmd
1.1 Inputs
Raw FastQ files with the following pattern:
*.fastq.gz
1.2 Outputs
The expected outputs will be:
multiqc_report.html: A summary report of the alignment results generated by MultiQC, in HTML format.*fastp-trim*.fq.gz: Trimmed FastQ files.Due to the large file sizes of FastQs, these cannot be hosted in the urol-e5/timeseries_molecular GitHub repo. As such these files are available for download here:
2 Create a Bash variables file
This allows usage of Bash variables across R Markdown chunks.
{
echo "#### Assign Variables ####"
echo ""
echo "# Data directories"
echo 'export repo_dir=/home/shared/8TB_HDD_01/sam/gitrepos/urol-e5/timeseries_molecular'
echo 'export output_dir_top=${repo_dir}/F-Ptua/output/01.00-F-Ptua-WGBS-trimming-fastp-FastQC-MultiQC'
echo 'export raw_reads_dir="${repo_dir}/F-Ptua/data/wgbs-raw-fastqs"'
echo 'export trimmed_fastqs_dir="${output_dir_top}"'
echo ""
echo "# Paths to programs"
echo 'export programs_dir="/home/shared"'
echo 'export bbmap_repair="${programs_dir}/bbmap-39.12/repair.sh"'
echo 'export bismark_dir="${programs_dir}/Bismark-0.24.0"'
echo 'export bowtie2_dir="${programs_dir}/bowtie2-2.4.4-linux-x86_64"'
echo 'export fastp="${programs_dir}/fastp-v0.24.0/fastp"'
echo 'export fastqc="${programs_dir}/FastQC-0.12.1/fastqc"'
echo 'export multiqc="/home/sam/programs/mambaforge/bin/multiqc"'
echo 'export samtools_dir="${programs_dir}/samtools-1.12"'
echo ""
echo "# Set FastQ filename patterns"
echo "export fastq_pattern='*.fastq.gz'"
echo "export R1_fastq_pattern='*_R1_*.fastq.gz'"
echo "export R2_fastq_pattern='*_R2_*.fastq.gz'"
echo "export trimmed_fastq_pattern='*fastp-trim*.fq.gz'"
echo ""
echo "# Set number of CPUs to use"
echo 'export threads=40'
echo ""
echo "## Inititalize arrays"
echo 'export fastq_array_R1=()'
echo 'export fastq_array_R2=()'
echo 'export trimmed_fastqs_array=()'
echo 'export R1_names_array=()'
echo 'export R2_names_array=()'
echo ""
echo "# Print formatting"
echo 'export line="--------------------------------------------------------"'
echo ""
} > .bashvars
cat .bashvars#### Assign Variables ####
# Data directories
export repo_dir=/home/shared/8TB_HDD_01/sam/gitrepos/urol-e5/timeseries_molecular
export output_dir_top=${repo_dir}/F-Ptua/output/01.00-F-Ptua-WGBS-trimming-fastp-FastQC-MultiQC
export raw_reads_dir="${repo_dir}/F-Ptua/data/wgbs-raw-fastqs"
export trimmed_fastqs_dir="${output_dir_top}"
# Paths to programs
export programs_dir="/home/shared"
export bbmap_repair="${programs_dir}/bbmap-39.12/repair.sh"
export bismark_dir="${programs_dir}/Bismark-0.24.0"
export bowtie2_dir="${programs_dir}/bowtie2-2.4.4-linux-x86_64"
export fastp="${programs_dir}/fastp-v0.24.0/fastp"
export fastqc="${programs_dir}/FastQC-0.12.1/fastqc"
export multiqc="/home/sam/programs/mambaforge/bin/multiqc"
export samtools_dir="${programs_dir}/samtools-1.12"
# Set FastQ filename patterns
export fastq_pattern='*.fastq.gz'
export R1_fastq_pattern='*_R1_*.fastq.gz'
export R2_fastq_pattern='*_R2_*.fastq.gz'
export trimmed_fastq_pattern='*fastp-trim*.fq.gz'
# Set number of CPUs to use
export threads=40
## Inititalize arrays
export fastq_array_R1=()
export fastq_array_R2=()
export trimmed_fastqs_array=()
export R1_names_array=()
export R2_names_array=()
# Print formatting
export line="--------------------------------------------------------"3 Trimming and Error Repair
Trimming will remove any Illumina sequencing adapters, as well as polyG and polyX (primarily polyA) sequences. Trimming will also remove the first 15bp from the 5’ end of each read - this is based on FastQC/MultiQC assessment of raw reads.
Samples generating an error during trimming will attempt to be repaired with BBTools’ repair.sh script.
# Load bash variables into memory
source .bashvars
# Make output directory, if it doesn't exist
mkdir --parents ${output_dir_top}
cd "${raw_reads_dir}"
# Create arrays of fastq R1 files and sample names
# Do NOT quote R1_fastq_pattern variable
for fastq in ${R1_fastq_pattern}
do
fastq_array_R1+=("${fastq}")
# Use parameter substitution to remove all text up to and including last "." from
# right side of string.
R1_names_array+=("${fastq%%.*}")
done
# Create array of fastq R2 files
# Do NOT quote R2_fastq_pattern variable
for fastq in ${R2_fastq_pattern}
do
fastq_array_R2+=("${fastq}")
# Use parameter substitution to remove all text up to and including last "." from
# right side of string.
R2_names_array+=(${fastq%%.*})
done
# Run fastp on files
# Adds JSON report output for downstream usage by MultiQC
echo "Beginning fastp trimming."
echo "-------------------------------------------------"
echo ""
for index in "${!fastq_array_R1[@]}"
do
R1_sample_name="${R1_names_array[index]}"
R2_sample_name="${R2_names_array[index]}"
stderr_PE_name=$(echo ${R1_sample_name} | awk -F"_" '{print $1}')
${fastp} \
--in1 ${fastq_array_R1[index]} \
--in2 ${fastq_array_R2[index]} \
--detect_adapter_for_pe \
--trim_poly_g \
--trim_poly_x \
--thread 16 \
--trim_front1 15 \
--trim_front2 15 \
--html ${output_dir_top}/"${R1_sample_name}".fastp-trim.report.html \
--json ${output_dir_top}/"${R1_sample_name}".fastp-trim.report.json \
--out1 ${output_dir_top}/"${R1_sample_name}".fastp-trim.fq.gz \
--out2 ${output_dir_top}/"${R2_sample_name}".fastp-trim.fq.gz \
2> ${output_dir_top}/"${stderr_PE_name}".fastp-trim.stderr
# Check for errors in fastp stderr
grep --before-context 5 "ERROR" ${output_dir_top}/"${stderr_PE_name}".fastp-trim.stderr
# Check for fastp errors and then repair
if grep --quiet "ERROR" ${output_dir_top}/"${stderr_PE_name}".fastp-trim.stderr; then
rm ${output_dir_top}/"${R1_sample_name}".fastp-trim.fq.gz
rm ${output_dir_top}/"${R2_sample_name}".fastp-trim.fq.gz
${bbmap_repair} \
in1=${fastq_array_R1[index]} \
in2=${fastq_array_R2[index]} \
out1="${R1_sample_name}".REPAIRED.fastq.gz \
out2="${R2_sample_name}".REPAIRED.fastq.gz \
outs=/dev/null \
2> "${R1_sample_name}".REPAIRED.stderr
${fastp} \
--in1 "${R1_sample_name}".REPAIRED.fastq.gz \
--in2 "${R2_sample_name}".REPAIRED.fastq.gz \
--detect_adapter_for_pe \
--trim_poly_g \
--trim_poly_x \
--thread 16 \
--trim_front1 10 \
--trim_front2 10 \
--html ${output_dir_top}/"${R1_sample_name}".fastp-trim.REPAIRED.report.html \
--json ${output_dir_top}/"${R1_sample_name}".fastp-trim.REPAIRED.report.json \
--out1 ${output_dir_top}/"${R1_sample_name}".fastp-trim.REPAIRED.fq.gz \
--out2 ${output_dir_top}/"${R2_sample_name}".fastp-trim.REPAIRED.fq.gz \
2> ${output_dir_top}/"${stderr_PE_name}".fastp-trim.REPAIRED.stderr
if grep --quiet "ERROR" ${output_dir_top}/"${stderr_PE_name}".fastp-trim.REPAIRED.stderr; then
echo "These ${stderr_PE_name} samples are broken."
echo "Just give up. :("
echo ""
fi
fi
echo "Finished trimming:"
echo "${fastq_array_R1[index]}"
echo "${fastq_array_R2[index]}"
echo ""
# Generate md5 checksums for newly trimmed files
cd "${output_dir_top}"
md5sum "${R1_sample_name}".fastp-trim.fq.gz > "${R1_sample_name}".fastp-trim.fq.gz.md5
md5sum "${R2_sample_name}".fastp-trim.fq.gz > "${R2_sample_name}".fastp-trim.fq.gz.md5
cd "${raw_reads_dir}"
doneBeginning fastp trimming.
-------------------------------------------------
Finished trimming:
POC-201-TP1_R1_001.fastq.gz
POC-201-TP1_R2_001.fastq.gz
Finished trimming:
POC-201-TP2_R1_001.fastq.gz
POC-201-TP2_R2_001.fastq.gz
Finished trimming:
POC-201-TP3_R1_001.fastq.gz
POC-201-TP3_R2_001.fastq.gz
Finished trimming:
POC-219-TP1_R1_001.fastq.gz
POC-219-TP1_R2_001.fastq.gz
Finished trimming:
POC-219-TP2_R1_001.fastq.gz
POC-219-TP2_R2_001.fastq.gz
Finished trimming:
POC-219-TP3_R1_001.fastq.gz
POC-219-TP3_R2_001.fastq.gz
Finished trimming:
POC-219-TP4_R1_001.fastq.gz
POC-219-TP4_R2_001.fastq.gz
Finished trimming:
POC-222-TP1_R1_001.fastq.gz
POC-222-TP1_R2_001.fastq.gz
Finished trimming:
POC-222-TP2_R1_001.fastq.gz
POC-222-TP2_R2_001.fastq.gz
Finished trimming:
POC-222-TP3_R1_001.fastq.gz
POC-222-TP3_R2_001.fastq.gz
Finished trimming:
POC-222-TP4_R1_001.fastq.gz
POC-222-TP4_R2_001.fastq.gz
Finished trimming:
POC-255-TP1_R1_001.fastq.gz
POC-255-TP1_R2_001.fastq.gz
Finished trimming:
POC-255-TP2_R1_001.fastq.gz
POC-255-TP2_R2_001.fastq.gz
Finished trimming:
POC-255-TP3_R1_001.fastq.gz
POC-255-TP3_R2_001.fastq.gz
Finished trimming:
POC-255-TP4_R1_001.fastq.gz
POC-255-TP4_R2_001.fastq.gz
Finished trimming:
POC-259-TP1_R1_001.fastq.gz
POC-259-TP1_R2_001.fastq.gz
Finished trimming:
POC-259-TP2_R1_001.fastq.gz
POC-259-TP2_R2_001.fastq.gz
Finished trimming:
POC-259-TP3_R1_001.fastq.gz
POC-259-TP3_R2_001.fastq.gz
Finished trimming:
POC-259-TP4_R1_001.fastq.gz
POC-259-TP4_R2_001.fastq.gz
Finished trimming:
POC-40-TP1_R1_001.fastq.gz
POC-40-TP1_R2_001.fastq.gz
Finished trimming:
POC-40-TP2_R1_001.fastq.gz
POC-40-TP2_R2_001.fastq.gz
Finished trimming:
POC-40-TP3_R1_001.fastq.gz
POC-40-TP3_R2_001.fastq.gz
Finished trimming:
POC-40-TP4_R1_001.fastq.gz
POC-40-TP4_R2_001.fastq.gz
Finished trimming:
POC-42-TP1_R1_001.fastq.gz
POC-42-TP1_R2_001.fastq.gz
Finished trimming:
POC-42-TP2_R1_001.fastq.gz
POC-42-TP2_R2_001.fastq.gz
Finished trimming:
POC-42-TP3_R1_001.fastq.gz
POC-42-TP3_R2_001.fastq.gz
Finished trimming:
POC-42-TP4_R1_001.fastq.gz
POC-42-TP4_R2_001.fastq.gz
Finished trimming:
POC-52-TP1_R1_001.fastq.gz
POC-52-TP1_R2_001.fastq.gz
Finished trimming:
POC-52-TP2_R1_001.fastq.gz
POC-52-TP2_R2_001.fastq.gz
Finished trimming:
POC-52-TP3_R1_001.fastq.gz
POC-52-TP3_R2_001.fastq.gz
Finished trimming:
POC-52-TP4_R1_001.fastq.gz
POC-52-TP4_R2_001.fastq.gz
Finished trimming:
POC-53-TP1_R1_001.fastq.gz
POC-53-TP1_R2_001.fastq.gz
Finished trimming:
POC-53-TP2_R1_001.fastq.gz
POC-53-TP2_R2_001.fastq.gz
Finished trimming:
POC-53-TP3_R1_001.fastq.gz
POC-53-TP3_R2_001.fastq.gz
Finished trimming:
POC-53-TP4_R1_001.fastq.gz
POC-53-TP4_R2_001.fastq.gz
Finished trimming:
POC-57-TP1_R1_001.fastq.gz
POC-57-TP1_R2_001.fastq.gz
Finished trimming:
POC-57-TP2_R1_001.fastq.gz
POC-57-TP2_R2_001.fastq.gz
Finished trimming:
POC-57-TP3_R1_001.fastq.gz
POC-57-TP3_R2_001.fastq.gz
Finished trimming:
POC-57-TP4_R1_001.fastq.gz
POC-57-TP4_R2_001.fastq.gz4 FastQC
# Load bash variables into memory
source .bashvars
cd "${output_dir_top}"
############ RUN FASTQC ############
# Create array of trimmed FastQs
trimmed_fastqs_array=(${trimmed_fastq_pattern})
# Pass array contents to new variable as space-delimited list
trimmed_fastqc_list=$(echo "${trimmed_fastqs_array[*]}")
echo "Beginning FastQC on trimmed reads..."
echo ""
# Run FastQC
### NOTE: Do NOT quote raw_fastqc_list
${fastqc} \
--threads ${threads} \
--outdir "${output_dir_top}" \
--quiet \
${trimmed_fastqc_list}
echo "FastQC on trimmed reads complete!"
echo ""
############ END FASTQC ############Beginning FastQC on trimmed reads...
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
application/gzip
FastQC on trimmed reads complete!5 MultiQC
Uses --cl-config "sp: { fastp: { fn: '*report.json' } }" to update the MultiQC search pattern for the fastp module.
# Load bash variables into memory
source .bashvars
cd "${output_dir_top}"
${multiqc} . \
--cl-config "sp: { fastp: { fn: '*report.json' } }" \
--interactive
# Remove zip files
rm *_fastqc.zip /// MultiQC 🔍 | v1.14
| multiqc | MultiQC Version v1.29 now available!
| multiqc | Search path : /home/shared/8TB_HDD_01/sam/gitrepos/urol-e5/timeseries_molecular/F-Ptua/output/01.00-F-Ptua-WGBS-trimming-fastp-FastQC-MultiQC
| searching | ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 429/429
| fastp | Found 39 reports
| fastqc | Found 78 reports
| multiqc | Compressing plot data
| multiqc | Report : multiqc_report.html
| multiqc | Data : multiqc_data
| multiqc | MultiQC completeRESULTS
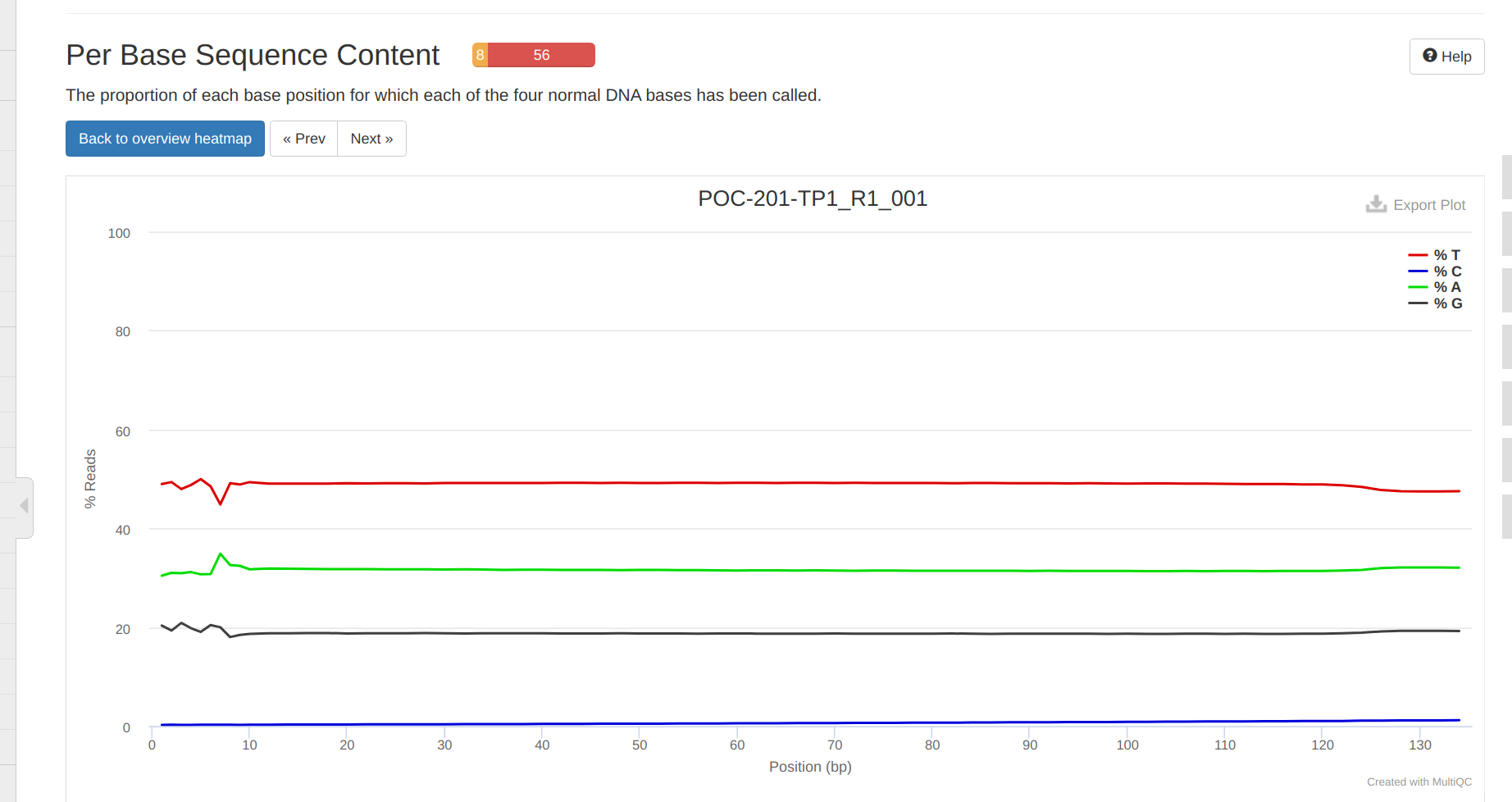
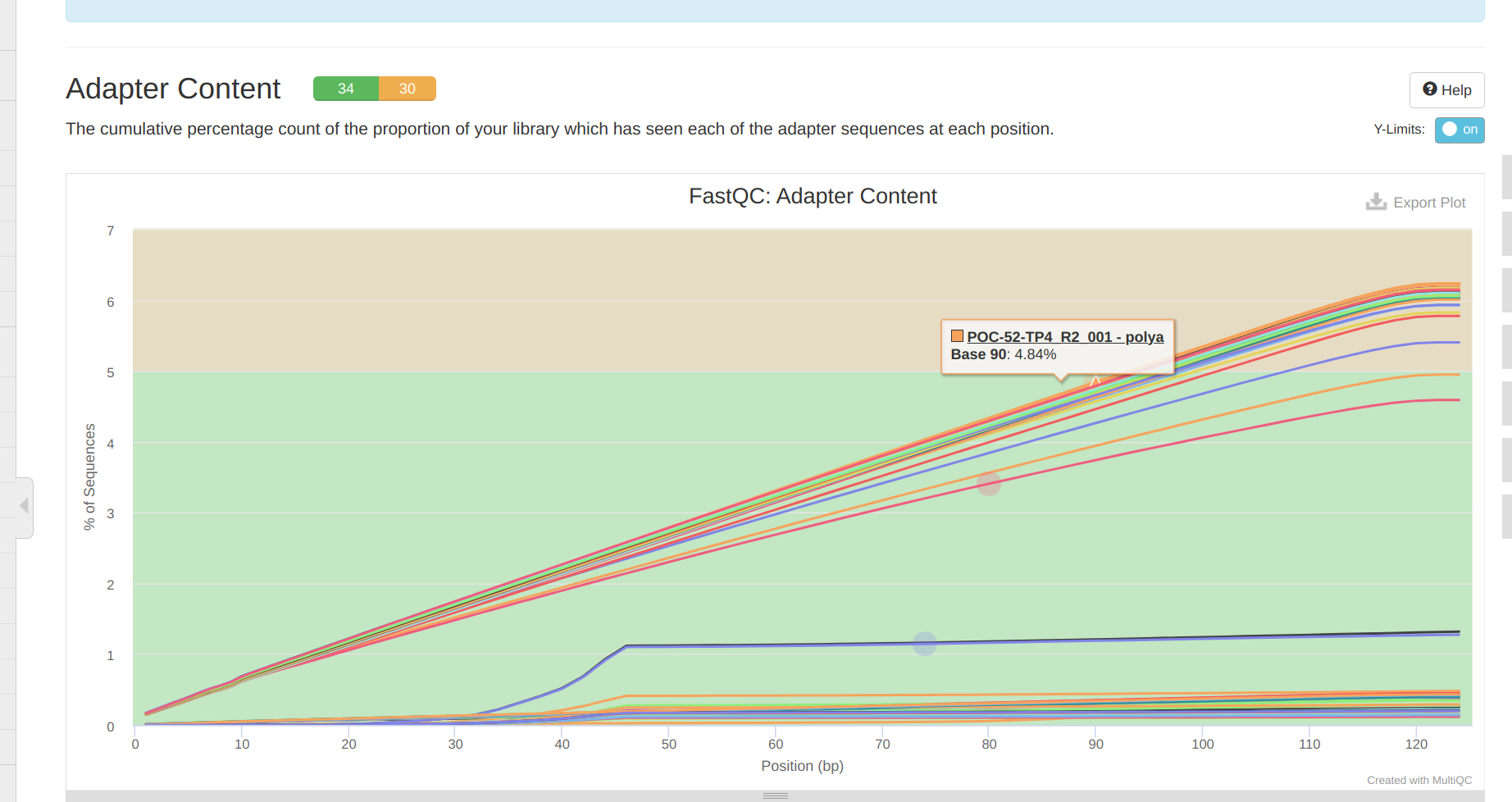
Overall, trimming looks okay. Surprisingly, the 5’ ends still exhibit some variation in the sequnce contant. Additionally,, it’s disappointing to see residual polyA contamination, despite specifying --trim_poly_x in the fastp arguments. The polyA contamination is likely RNA carried over from the DNA isolation prep. It shouldn’t cause any issues, as RNA shouldn’t end up aligning to the bisulfite-converted genome downstream.Add commentMore actions
MultiQC report: