In another troubleshooting attempt for this problematic BS-seq Illumina data, I’m going to use Trimmomatic to remove the first 39 bases of each read. This is due to the fact that even after the previous quality trimming with Trimmomatic, the first 39 bases still showed inconsistent quality:
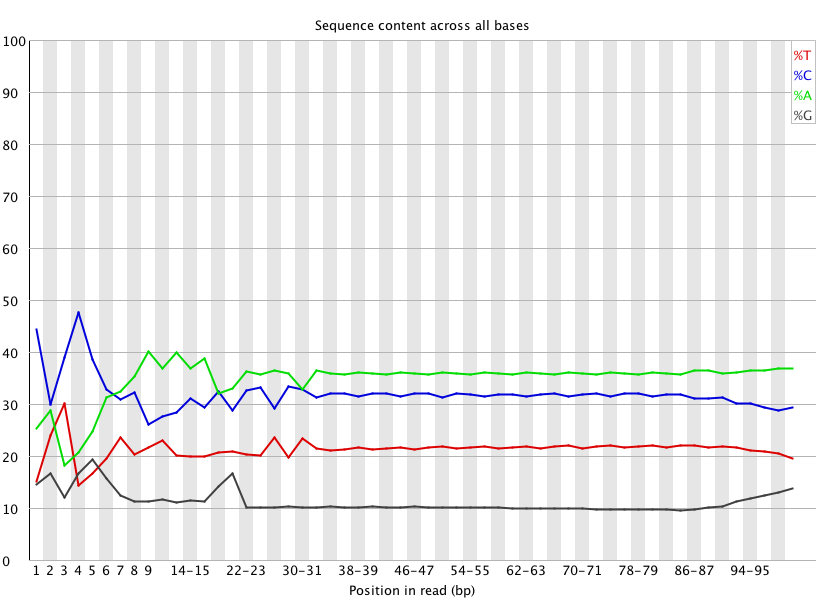 (http://eagle.fish.washington.edu/Arabidopsis/20150414_trimmed_2212_lane2_CTTGTA_L002_R1_001_fastqc/Images/per_base_sequence_content.png)
(http://eagle.fish.washington.edu/Arabidopsis/20150414_trimmed_2212_lane2_CTTGTA_L002_R1_001_fastqc/Images/per_base_sequence_content.png)
Ran Trimmomatic on just a single data set to try things out: 2212_lane2_CTTGTA_L002_R1_001.fastq.gz
Notebook Viewer: 20150506_Cgigas_larvae_OA_trimmomatic_FASTQC
Jupyter (IPython) notebook: 20150506_Cgigas_larvae_OA_trimmomatic_FASTQC.ipynb
Results:
Trimmed FASTQ: 20150506_trimmed_2212_lane2_CTTGTA_L002_R1_001.fastq.gz
FASTQC Report: 20150506_trimmed_2212_lane2_CTTGTA_L002_R1_001_fastqc.html
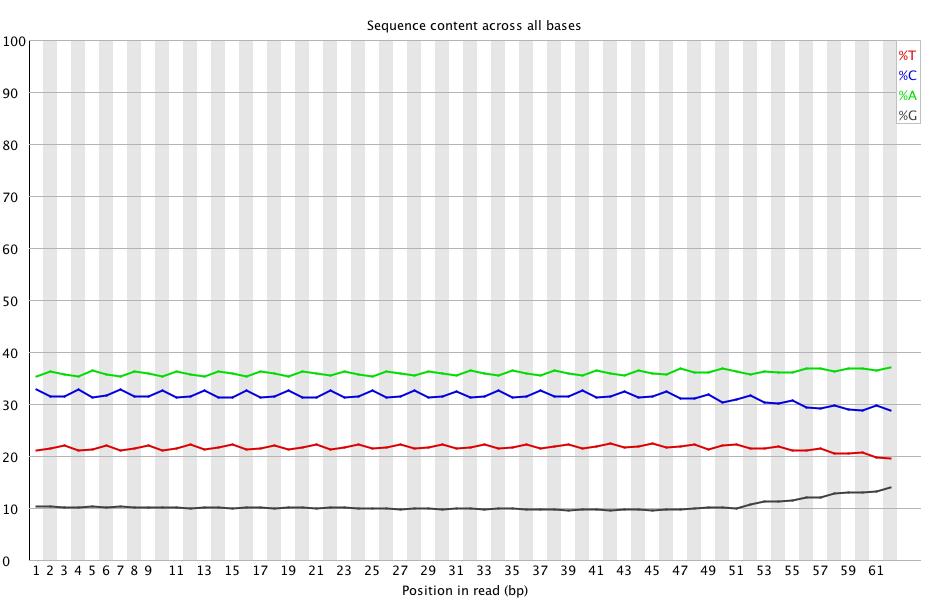 (http://eagle.fish.washington.edu/Arabidopsis/20150506_trimmed_2212_lane2_CTTGTA_L002_R1_001_fastqc/Images/per_base_sequence_content.png)
(http://eagle.fish.washington.edu/Arabidopsis/20150506_trimmed_2212_lane2_CTTGTA_L002_R1_001_fastqc/Images/per_base_sequence_content.png)
You can see how flat the newly trimmed data is (which is what one would expect).
Steven will take this trimmed dataset and try additional mapping with it to see if removal of the first 39 bases will improve the mapping.