INTRO
The original _Salvelinus namaycush C1q bisulfite PCR on 20240911 (Notebook) using bisulfite primers designed on 20240729 (Notebook) failed to produce any amplification. In response, we opted to just confirm that we could actually amplify the C1q gene using regular primers, designed on 20240916 (Notebook) and the original liver gDNA from 20240712. This latter PCR was successful. So, just for kicks, we decided to try running a PCR with the regular C1q primers and the bisulfite-treated gDNA.
MATERIALS & METHODS
DNA Pooling
Used 0.5uL of the pooled gDNA in each reaction.
PCR Reactions
PCR reactions were run using EpiMark Hot Start Taq DNA Polymerase (New England Biolabs), according to the manufacturer’s protocol, in a total reaction volume of 25uL.
I ran the reaction in duplicate, using 0.5uL of the pooled liver gDNA for template. Each reaction was prepared individually (i.e. no master mix was created).
Primers used:
Sn-C1q-F/R(SRIDs: 1842/3)
PCR Cycling
Annealing temperature gradient PCR was performed in a CFX Connect (BioRad) thermalcycler.
Cycling params:
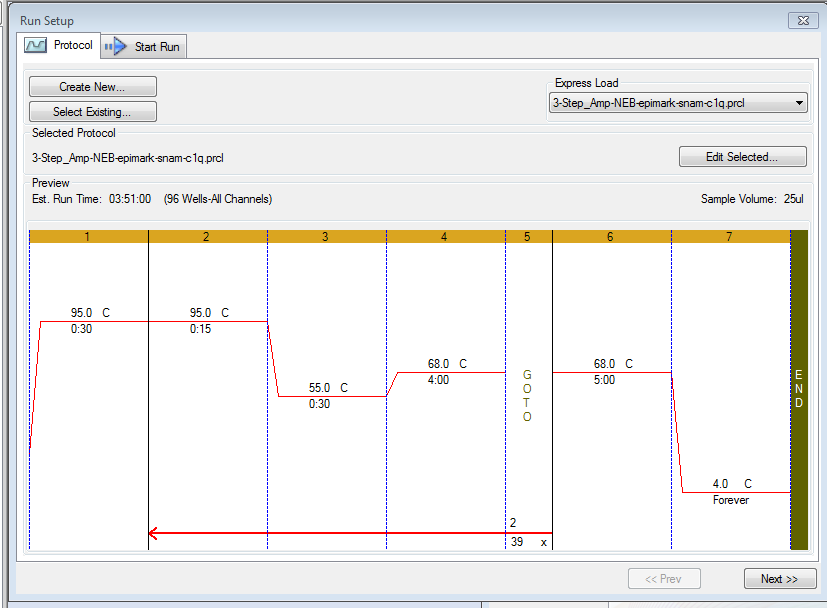
- 95oC - 30s
- 95oC - 15s
- 55oC - 30s
- 68oC - 4m
- Go to Step 2 x 39.
- 68oC - 5m
- 4oC - FOREVER
Extension step was set to 4m due to the expected amplicon size of C1q using the Roberts Lab primers (Notebook) of 3192bp. Most DNA polymerases (including EpiMark Hot Start Taq DNA Polymerase) process 1Kbp/min, so setting to four mins is a tad long, but will certainly ensure the full gene length is amplified.
PCR reactions were loaded on a low-TAE, 1.0% agarose gel, with ethidium bromide. Gel was run at 107V for ~45mins and then imaged.
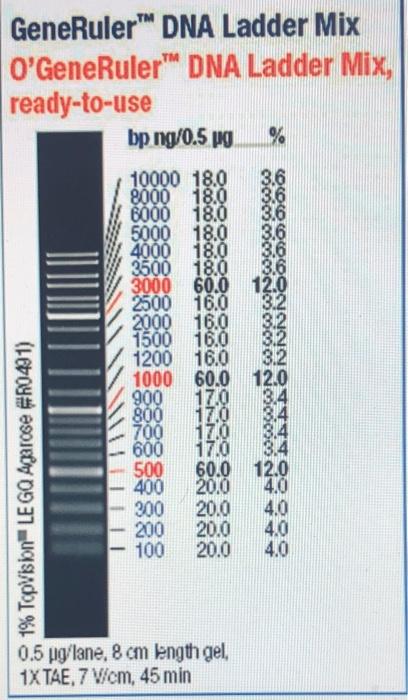
O’GeneRuler (ThermoFisher) Ladder was used (5uL) for size reference:
RESULTS
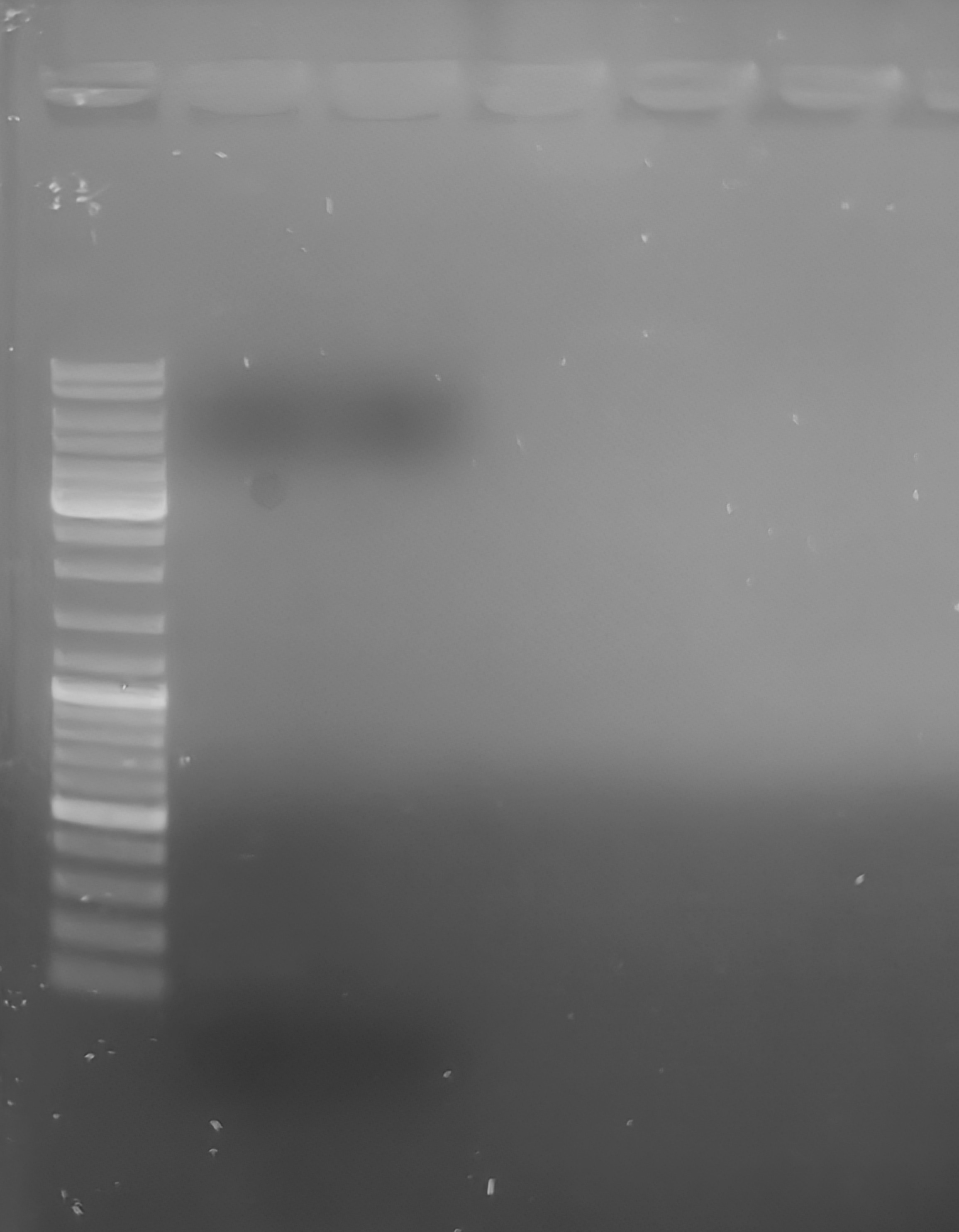
Unsurprisingly, we do not see any amplification.
DISCUSSION
Based on the failure of our previous bisulfite PCR using bisulfite primers (Notebook), which utilized primers specifically designed to amplify bisulfite-treated DNA, I had little expectation this would work. It did not.
In retrospect, we probably shouldn’t have had any expectations this would work. The reasons for this are related to the amount of DNA fragmentation/degradation that takes place during bisulfite treatment of gDNA. I was certainly aware of the fact that bisulfite treatement was considered “harsh” and resulted in single-stranded gDNA, as well as some degradation. However, after some investigation into this (spurred by the results of this project), it’s clear that the degradation that occurs is high! High enough, in fact, one is unlikely to ever expect any successful PCR of products larger than ~2,000bp.
As an example, ZymoResearch’s “Bisulfite Beginner Guide” (ZymoResearch website), provides an example gel to display the level of degradation expected from bisulfite treatment of gDNA. Keep in mind, companies usually display their best examples:

Notice that the molecular weight marker is a 100bp marker. This means the highest molecular weight band in the marker corresponds to 1500bp! That means our attempts to potentially amplify a ~3,200bp from bisulfite-treated gDNA were virtually impossible to achieve.
Also, as part of my research into the impacts of bisulfite degradation on gDNA, I came acroos this excellent paper (“Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR”) (Kint et al. 2018) which compares the performance of multiple bisulfite treatment kits, including:
- recovery (yield)
- recovery integrity
- processing time
The combination of information from that ZymoResearch site and the paper make it pretty clear that PCR of a product size of ~3,200bp would not happen. Have discussed with Steven and we’ll try to come up with some other ideas on how to approach the issue of determining gene-level DNA methylation of C1q in individuals.