INTRO
After a previous attempt at amplifying the C1q gene in bisulfite-converted DNA failed, we decided we should test out a standard PCR on untreated liver gDNA. I designed PCR primers for the C1Q gene on 20240916. I opted to test these out on a pool of the Salvelinus namaycush gDNA isolated on 20240712.
I also opted to test out some C1q gene primers I found in a box of Rick Goetz’s. My guess is that these are for qPCR, but since I’m testing gDNA and I don’t know the full gene structure (without needing to take the time to look), I decided to just try these as well.
MATERIALS & METHODS
DNA Pooling
1uL of each sample was used to create a pool of gDNA.
PCR Reactions
PCR reactions were run using EpiMark Hot Start Taq DNA Polymerase (New England Biolabs), according to the manufacturer’s protocol, in a total reaction volume of 25uL.
I ran each primer set in duplicate, using 0.5uL of the pooled liver gDNA for template. Each reaction was prepared individually (i.e. no master mix was created).
Primers used:
Sn-C1q-F/R(SRIDs: 1842/3)C1Q F.1/2(Rick Goetz primers)

PCR Cycling
Annealing temperature gradient PCR was performed in a CFX Connect (BioRad) thermalcycler.
Cycling params:

- 95oC - 30s
- 95oC - 15s
- 55oC - 30s
- 68oC - 4m
- Go to Step 2 x 39.
- 68oC - 5m
- 4oC - FOREVER
Extension step was set to 4m due to the expected amplicon size of C1q using the Roberts Lab primers (Notebook) of 3192bp. Most DNA polymerases (including EpiMark Hot Start Taq DNA Polymerase) process 1Kbp/min, so setting to four mins is a tad long, but will certainly ensure the full gene length is amplified.
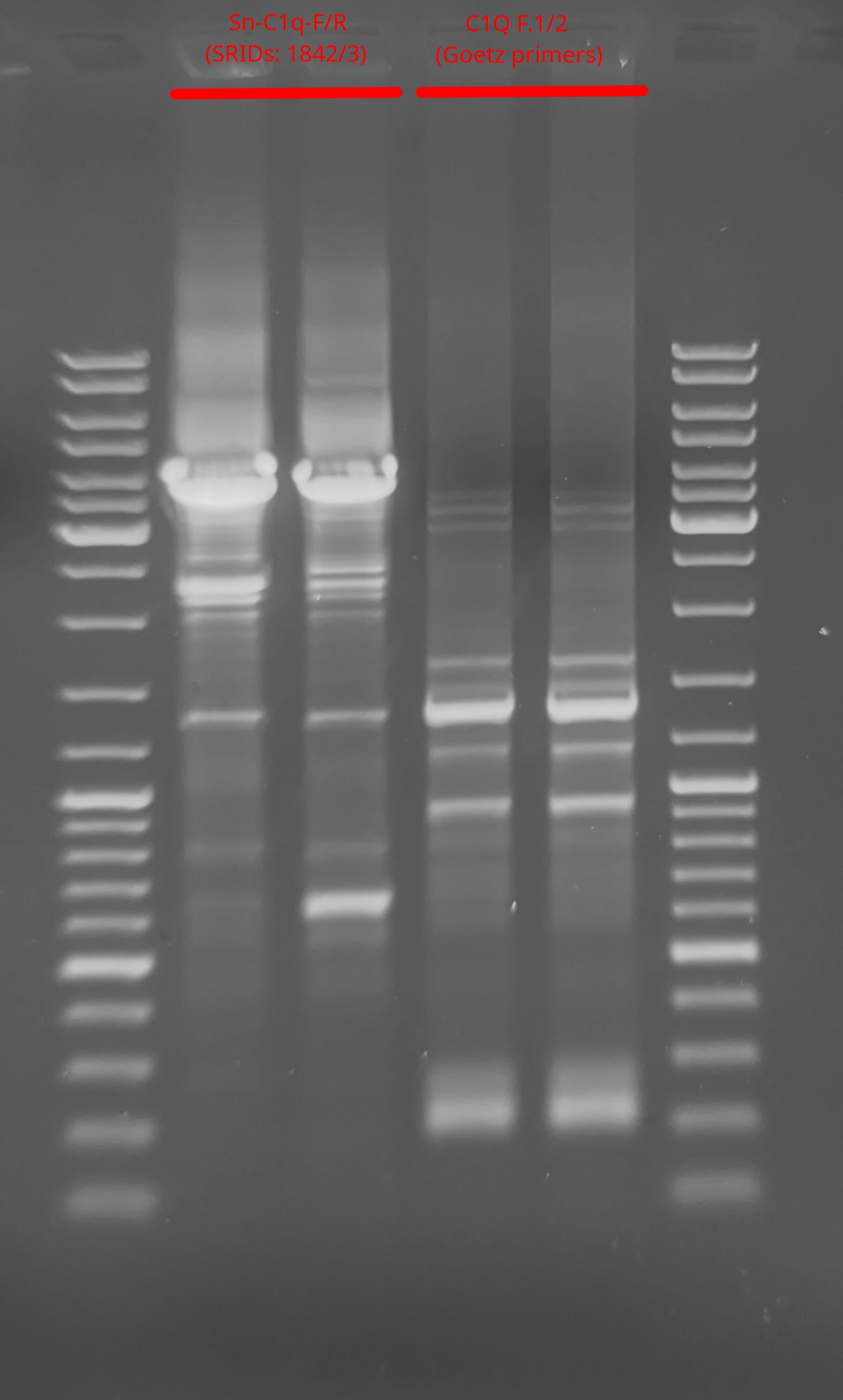
PCR reactions were loaded on a low-TAE, 1.0% agarose gel, with ethidium bromide. Gel was run at 107V for ~45mins and then imaged.
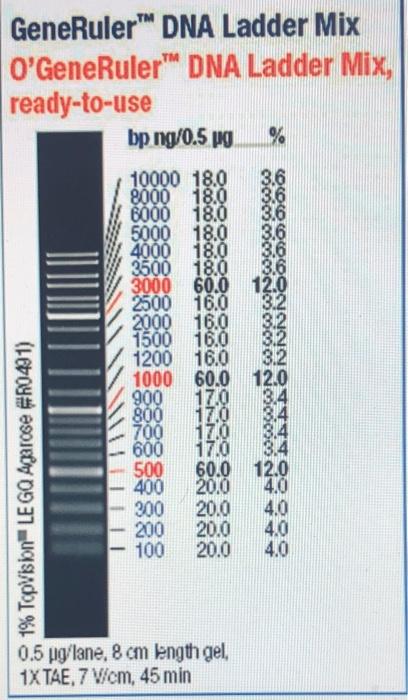
O’GeneRuler (ThermoFisher) Ladder was used (5uL) for size reference:
RESULTS
For the Sn-C1q-F/R (SRIDs: 1842/3) primers, we see a prominent amplicon at the expect size of ~3100bp.
With the C1Q F.1/2 (Rick Goetz primers), the most prominent band is ~1300bp. There’s nothing terribly informative about this, as these were just used a secondary means to confirm that the PCRs were working/failing.
Surprisingly, we also see a great deal of non-specific amplification. Although this is likely related to the annealing temp (55oC), it’s still surprising considering that I checked primer specificity using EMBOSS PrimeSearch (Notebook) and the primers each only had a single match in the genome…
DISCUSSION
These PCRs confirmed that the source DNA is good and primers designed using our methodology appears to work. To continue trying to address what’s wrong with the bisulfite primers/PCRs (Notebook), I’ll attempt to quantify the bisulfite-converted DNA (Notebook), which I did not do at the time. I’ll also run a PCR using the same primers used here (i.e. regular primers, not bisulfite primers) on the bisulfite-converted DNA, just to see what happens.