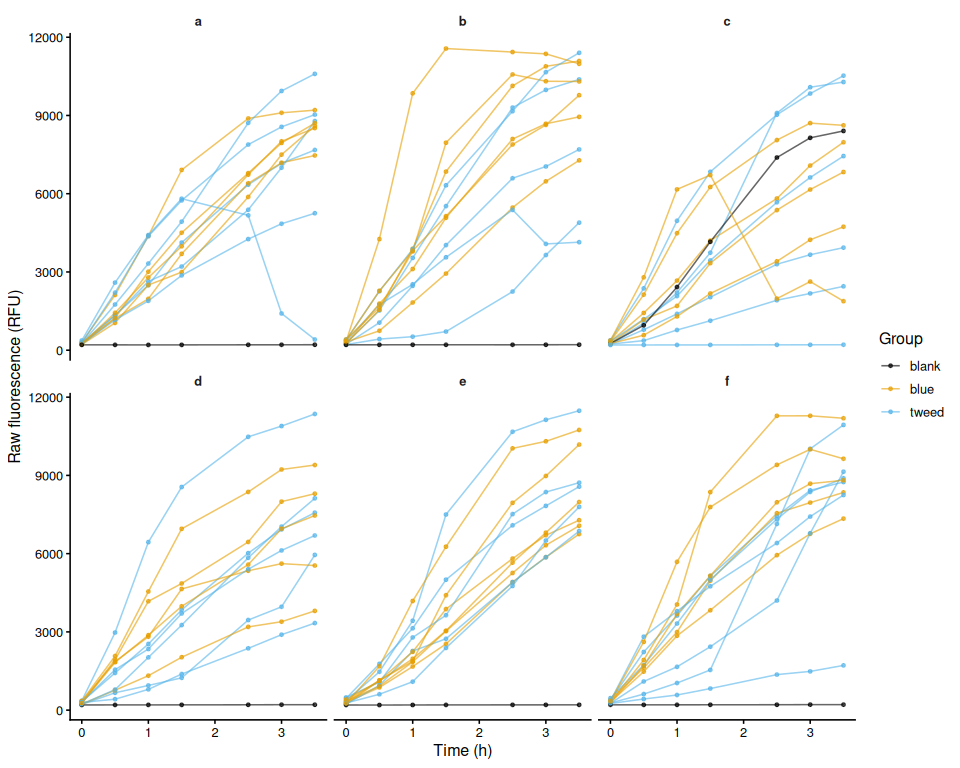
Homogenization - June 2026 SORMI M.gigas Ctenidia from Families 1 and 9 for Glycogen Glo Assay
2026
SORMI
Magallana gigas
ctenidia
Pacific oyster
Crassostrea gigas
glycogen
Glycogen Glo
homogenization

Resazurin Assays - USDA M.gigas Families in Response to Temperature Stress
2026
resazurin
Magallana gigas
Crassostrea gigas
Pacific oyster
heat stress
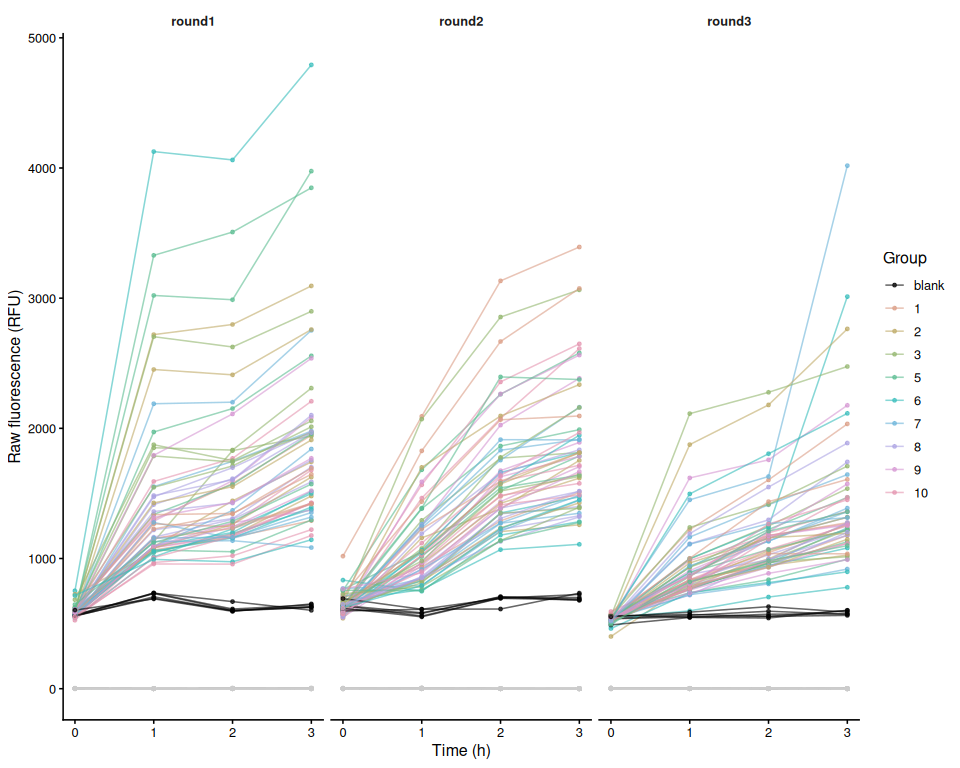
Resazurin Assays - USDA M.gigas Families in Response to Temperature Stress
2026
resazurin
Magallana gigas
Crassostrea gigas
Pacific oyster
heat stress
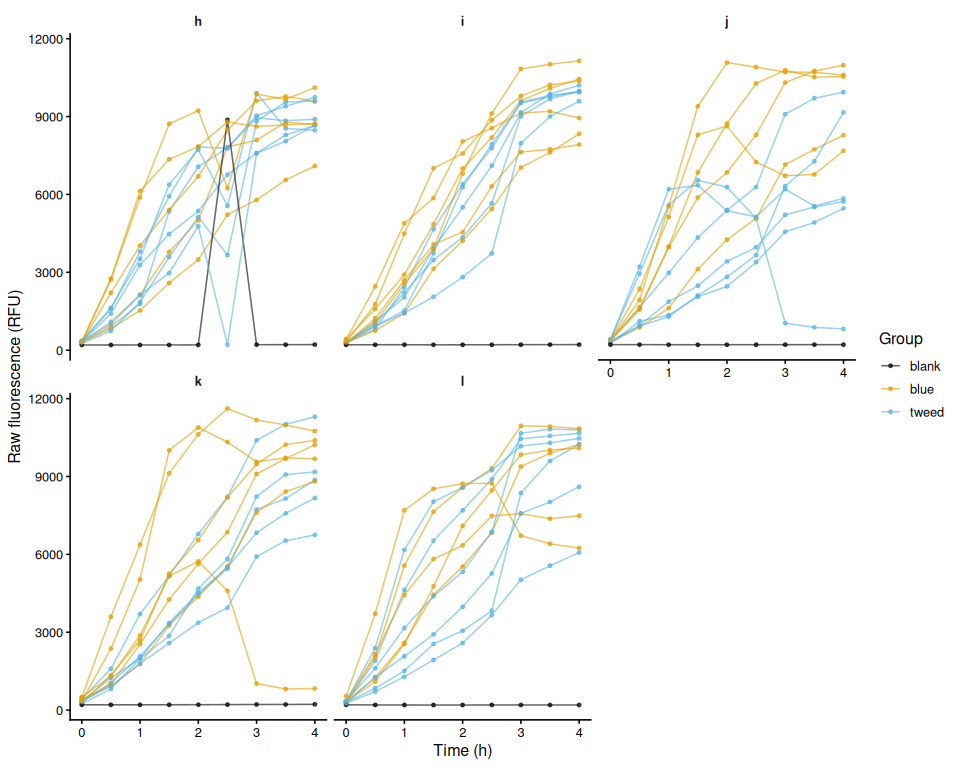
Heat Stress Survivorship - USDA M.gigas Families in Heat Stressed at 36C
2026
USDA
Pacific oysters
Crassostrea gigas
Magallana gigas
heat stress
survivorship
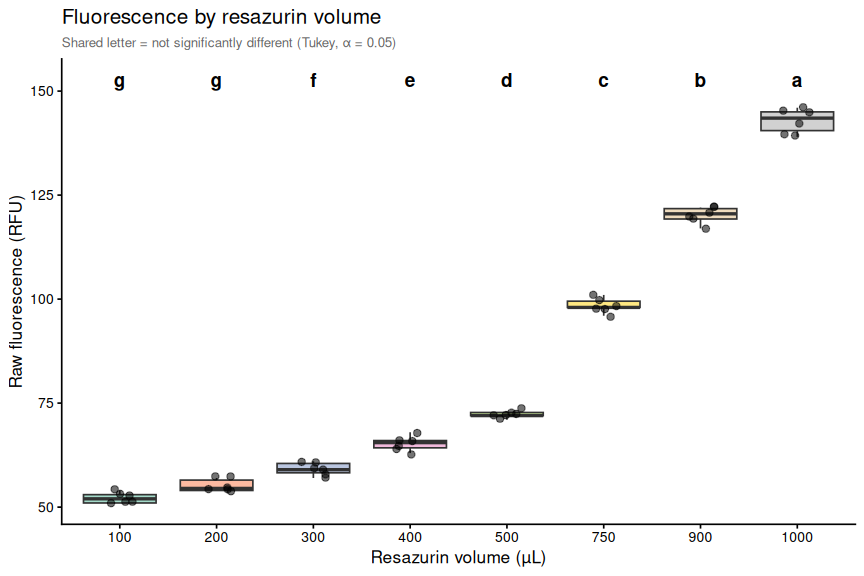
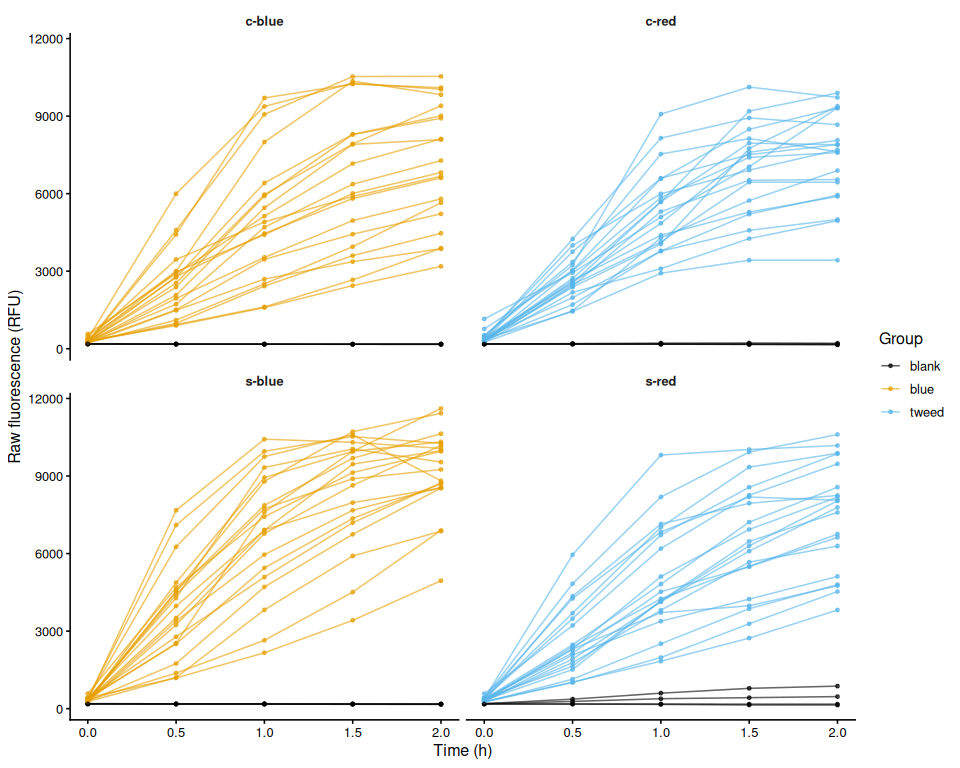
Resazurin Overview - Pacific Oyster USDA Heat Stress Raw Fluorescence
2026
SORMI
Pacific oyster
USDA
Crassostrea gigas
Magellana gigas
Resazurin Overview - Pacific oyster USDA Freshwater Stress Raw Fluorescence
2026
SORMI
Pacific oyster
USDA
Crassostrea gigas
Magellana gigas
Resazurin Assays - USDA M.gigas Families in Response to Temperature Stress
2026
Pacific oyster
resazurin
heat stress
Magallana gigas
Resazurin Assays - USDA M.gigas Families in Response to Freshwater Stress
2026
Pacific oyster
resazurin
heat stress
Magallana gigas
Crassostrea gigas
Resazurin Assays - USDA M.gigas Families in Response to Temperature Stress
2026
Pacific oyster
resazurin
heat stress
Magallana gigas
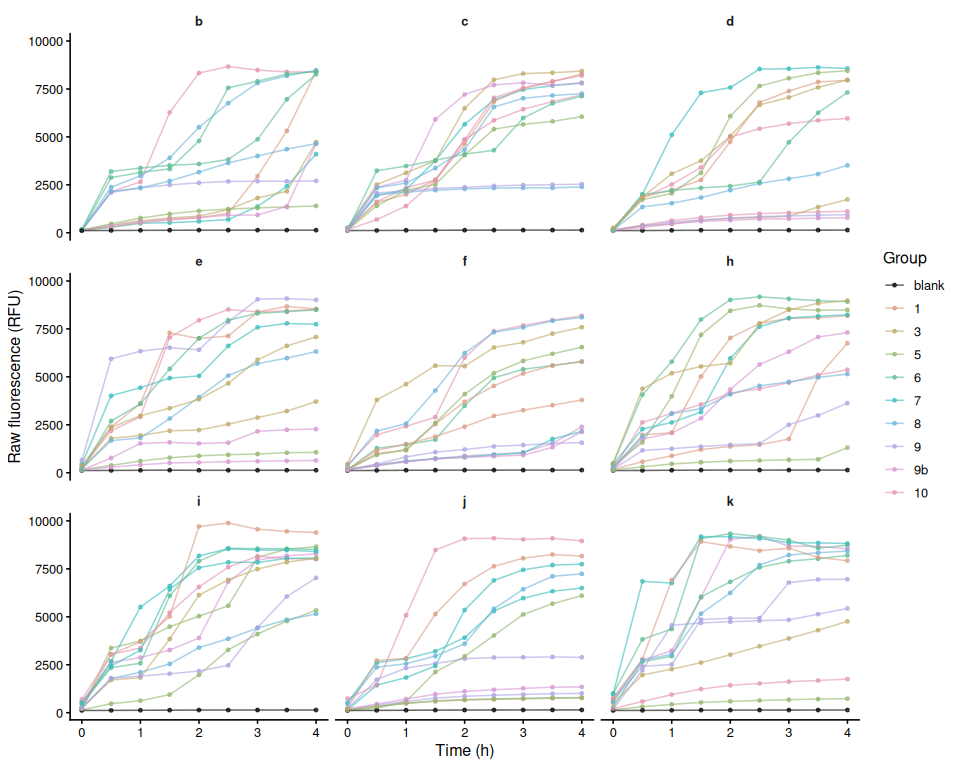
Resazurin Assays - Low Salinity Selected Juvenile Ruditapes phillippinarum Exposed to 36C Acute Heat Stress
2026
Ruditapes phillippinarum
heat stress
resazurin
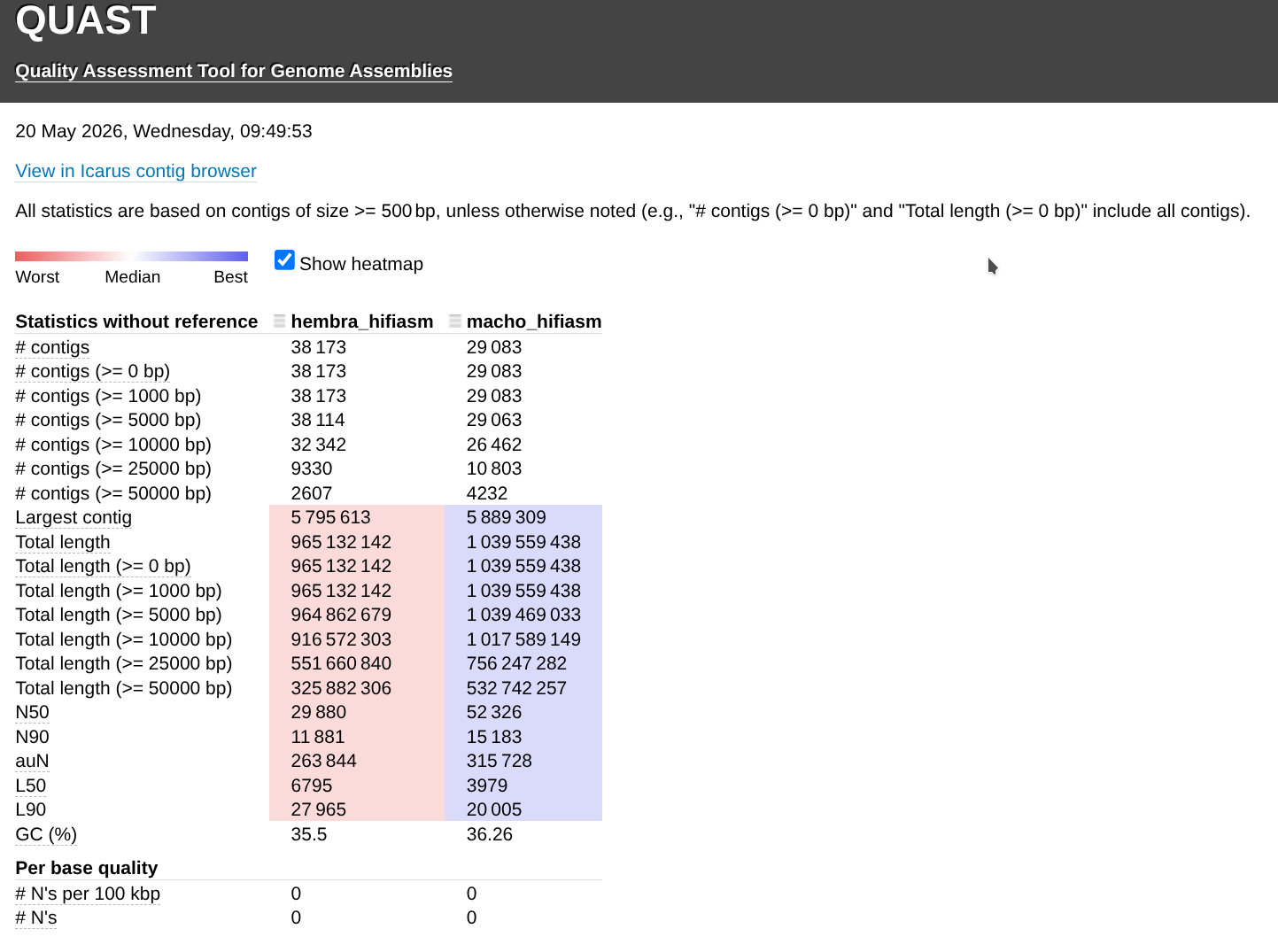
Genome Comparisons - C.rogercresseyi Hembra and Macho PacBio hifiasm Assemblies Using QUAST on Hyak
2026
hyak
Caligus rogercresseyi
QUAST
sea lice
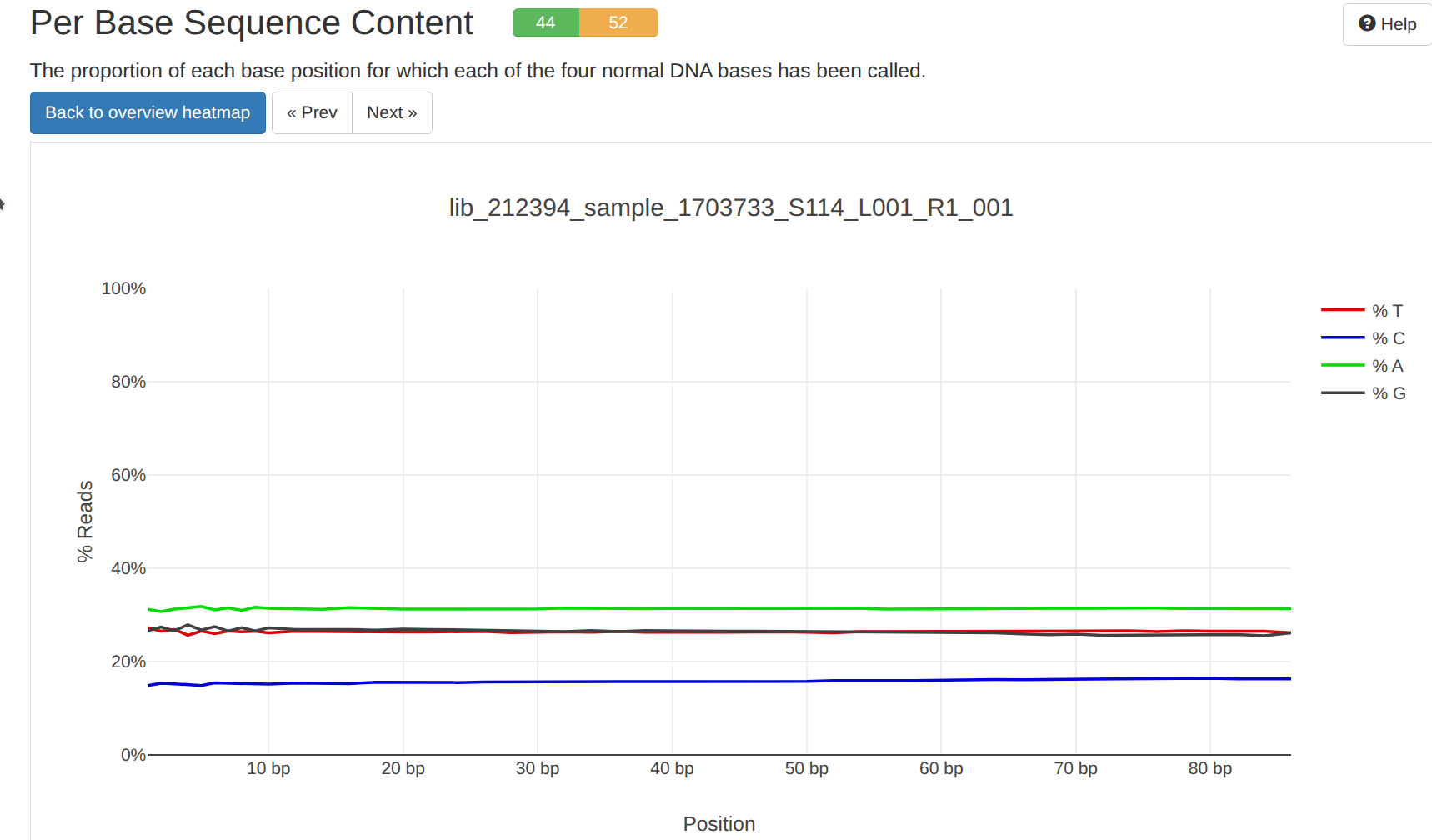
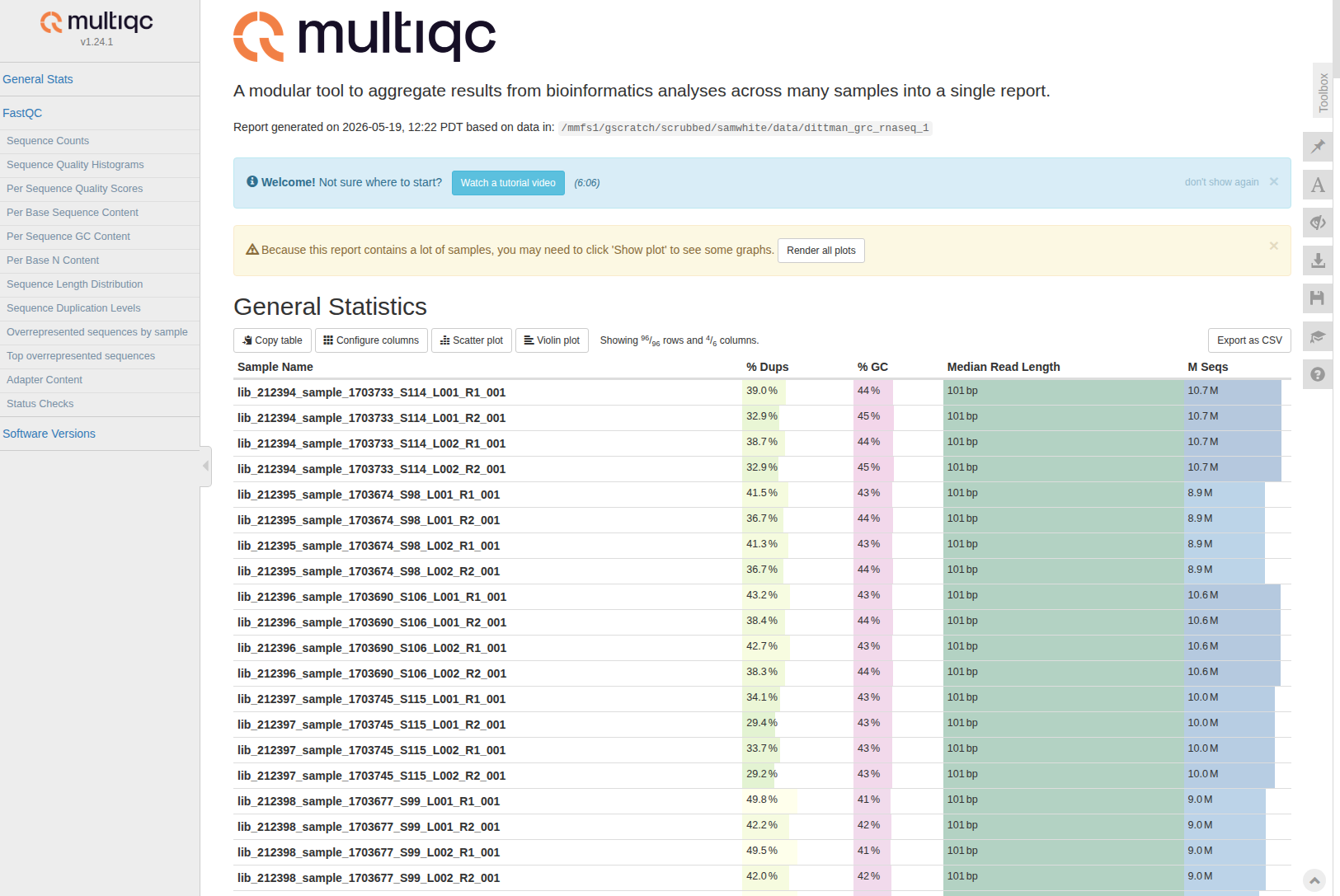
Trimming - Andy Dittman Preliminary RNA-seq Data Using Fastp FastQC MultiQC on Hyak
2026
fastp
fastqc
multiqc
hyak
RNA-seq
trimming
Genome Assembly - C.rogercresseyi Hembra with PacBio HiFi Reads Using hifiasm on Hyak
2026
sea lice
hyak
Caligus rogercresseyi
PacBio
hifiasm
genome assembly
Genome Assembly - C.rogercresseyi Macho with PacBio HiFi Reads Using hifiasm on Hyak
2026
sea lice
hyak
Caligus rogercresseyi
PacBio
hifiasm
genome assembly
Resazurin Summary - Manila Clam Heat Stress Experiments
2026
Ruditapes philippinarum
heat stress
resazurin
Manila clam
Heat Stress Survivorship - M.gigas USDA Families Heat Stressed at 33C
2026
USDA
Pacific oysters
Crassostrea gigas
Magallana gigas
heat stress
survivorship
Resazurin Assays - USDA M.gigas Families in Response to Freshwater Stress
2026
Pacific oyster
resazurin
freshwater stress
Magallana gigas
Crassostrea gigas
Data Received - Sea Lice PacBio Genome Sequencing
2026
Data Received
PacBio
Caligus rogercresseyi
sea lice
Genome Sequencing
Samples Received - P.generosa Gonad Histology Blocks and Slides
2026
Panopea generosa
Samples Received
gonad
histology
Pacific geoduck
Resazurin Assays - USDA M.gigas Families in Response to 36C and Freshwater Co-stressors
2026
Pacific oyster
resazurin
freshwater stress
heat stress
Magallana gigas
Crassostrea gigas
Resazurin Assays - USDA M.gigas Families in Response to Freshwater Stress
2026
Pacific oyster
resazurin
freshwater stress
Magallana gigas
Crassostrea gigas
Resazurin Assays - USDA M.gigas Families in Response to Freshwater Stress
2026
Pacific oyster
resazurin
freshwater stress
Magallana gigas
Crassostrea gigas
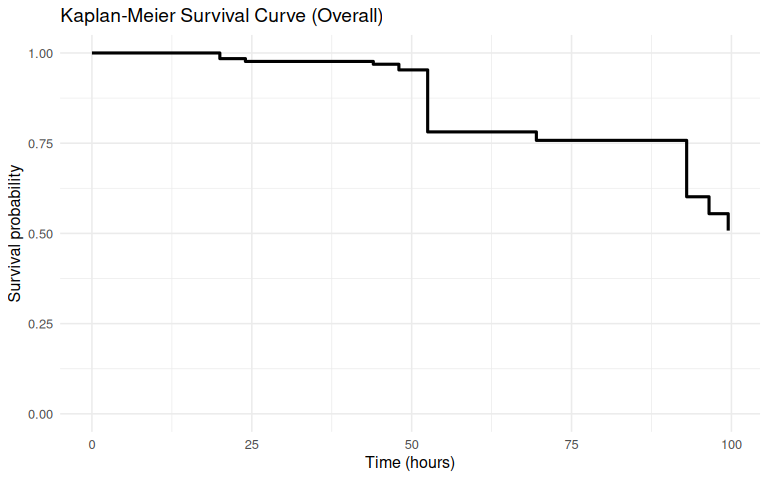
Heat Stress Survivorship - M.gigas USDA Families Heat Stressed at 33C
2026
USDA
Pacific oysters
Crassostrea gigas
Magallana gigas
heat stress
survivorship
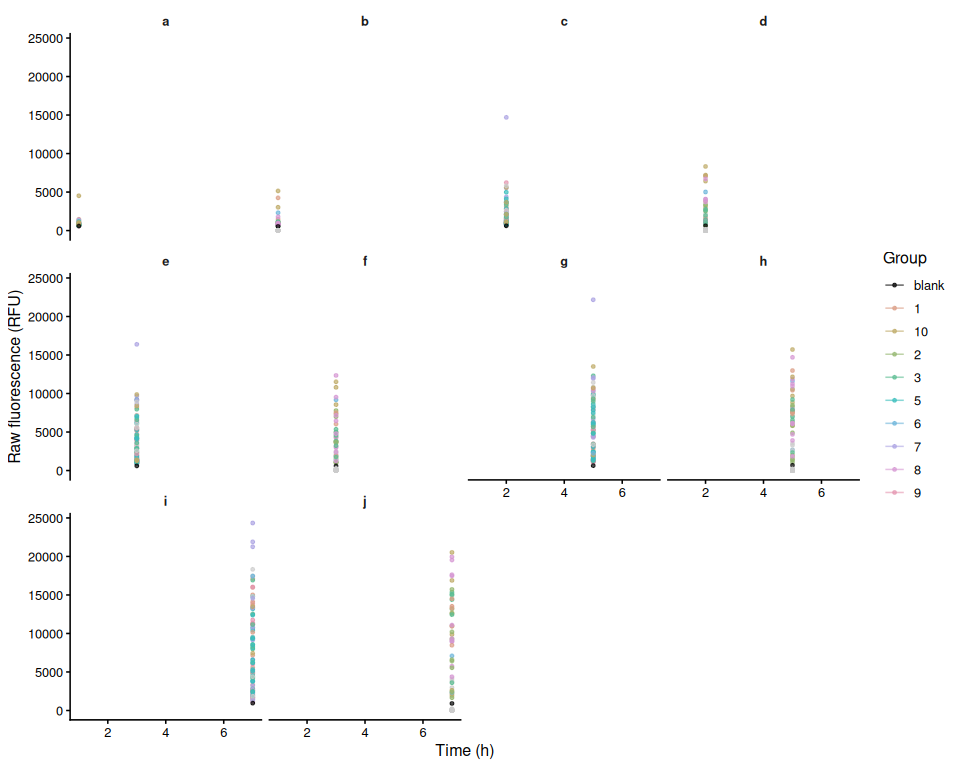
Resazurin Assays - USDA M.gigas Families in Response to Temperature Stress
2026
Resazurin
Pacific oyster
Crassostrea gigas
Magellana gigas

Samples Submitted - Geoduck Gonad Histology
2026
Pacific geoduck
Panopea generosa
histology
gonad
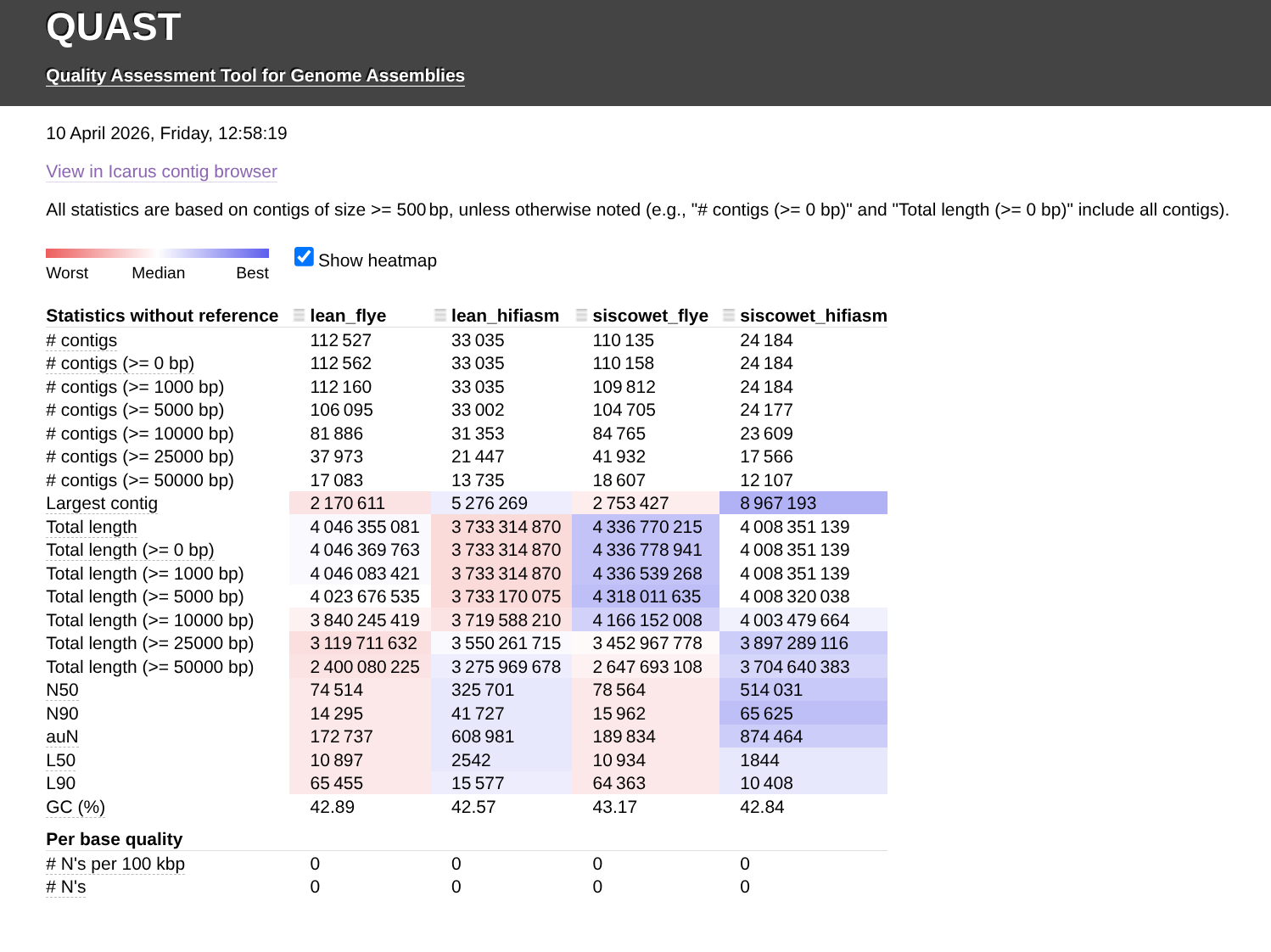
Genome Comparisons - Lake Trout PacBio Assemblies Flye vs hifiasm Using QUAST
2026
QUAST
flye
hifiasm
lake trout
genome assembly
PacBio
Salvelinus namaycush
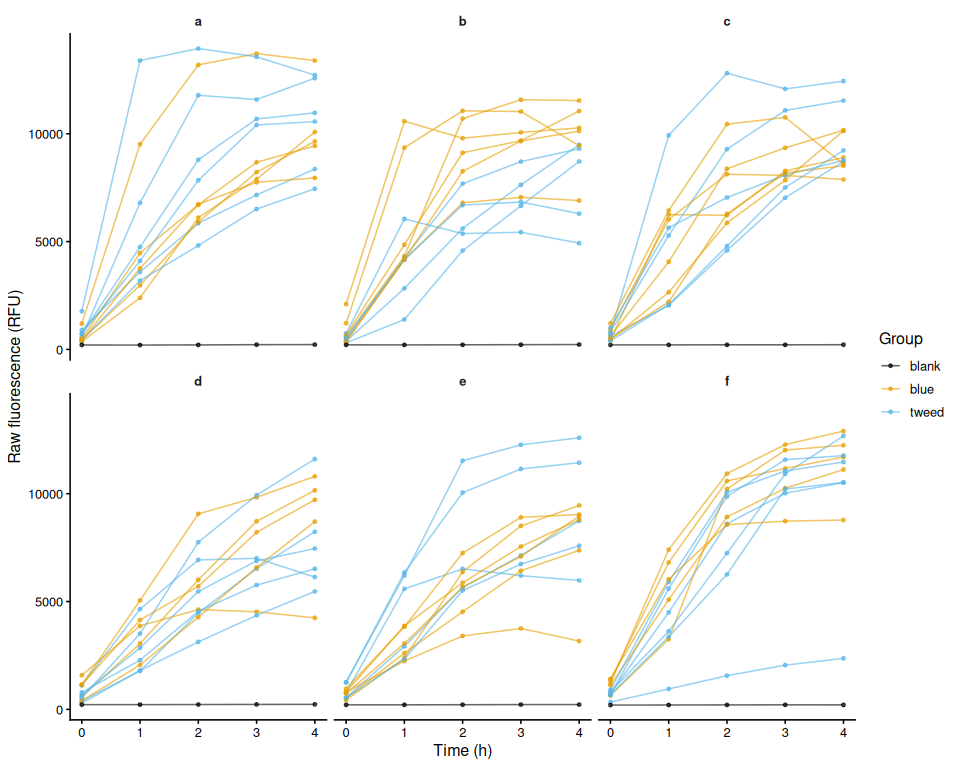
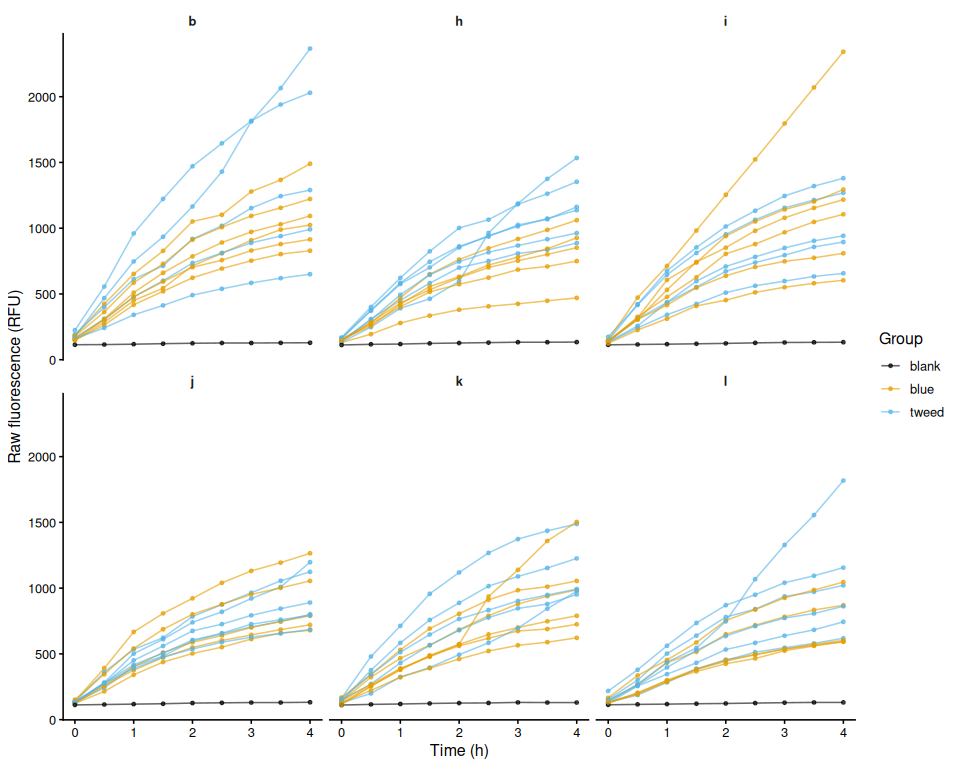
Resazurin Assays - Selected Juvenile Ruditapes philippinarum Exposed to 36C Acute Heat Stress Day 1
2026
resazurin
Ruditapes philippinarum
Manila clam
Synergy HTX
Resazurin Assays - Selected Juvenile Ruditapes philippinarum Exposed to 36C Acute Heat Stress
2026
resazurin
Ruditapes philippinarum
Manila clam
Synergy HTX
Genome Assembly - S.namaycush Lean Ecotype with PacBio HiFi Reads Using hifiasm on Hyak
2026
Genome Assembly
PacBio
HiFi Reads
hifiasm
Salvelinus namaycush
Lake Trout
Lean
Klone
Hyak
Genome Assembly - S.namaycush Siscowet Ecotype with PacBio HiFi Reads Using hifiasm on Hyak
2026
Genome Assembly
PacBio
HiFi Reads
hifiasm
Salvelinus namaycush
Lake Trout
Siscowet
Klone
Hyak
Resazurin Assays - Selected Juvenile Ruditapes philippinarum Exposed to 36C Acute Heat Stress Day 0
2026
resazurin
Ruditapes philippinarum
Manila clam
Synergy HTX
Resazurin Assays - Selected Juvenile Ruditapes philippinarum Exposed to 40C Acute Heat Stress
2026
resazurin
Ruditapes philippinarum
Manila clam
Synergy HTX
Genome Assembly - S.namaycush Lean Ecotype with PacBio HiFi Reads Using Flye
2026
Genome Assembly
PacBio
HiFi Reads
Flye
Salvelinus namaycush
Lake Trout
Lean
Genome Assembly - S.namaycush Siscowet Ecotype with PacBio HiFi Reads Using Flye
2026
Genome Assembly
PacBio
HiFi Reads
Flye
Salvelinus namaycush
Lake Trout
Siscowet

De Novo Transcriptome Assembly and Annotation - E5 A.pulchra RNA-seq Using Trinity and PASA
2026
Trinity
E5
PASA
transcriptome assembly
Acropora pulchra

De Novo Transcriptome Assembly and Annotation - E5 P.evermanni RNA-seq Using Trinity and PASA
2026
Trinity
E5
PASA
transcriptome assembly
Porites evermanni

De Novo Transcriptome Assembly and Annotation - E5 P.tuahiensis RNA-seq Using Trinity and PASA
2026
Trinity
E5
PASA
transcriptome assembly
Pocillopora tuahiensis
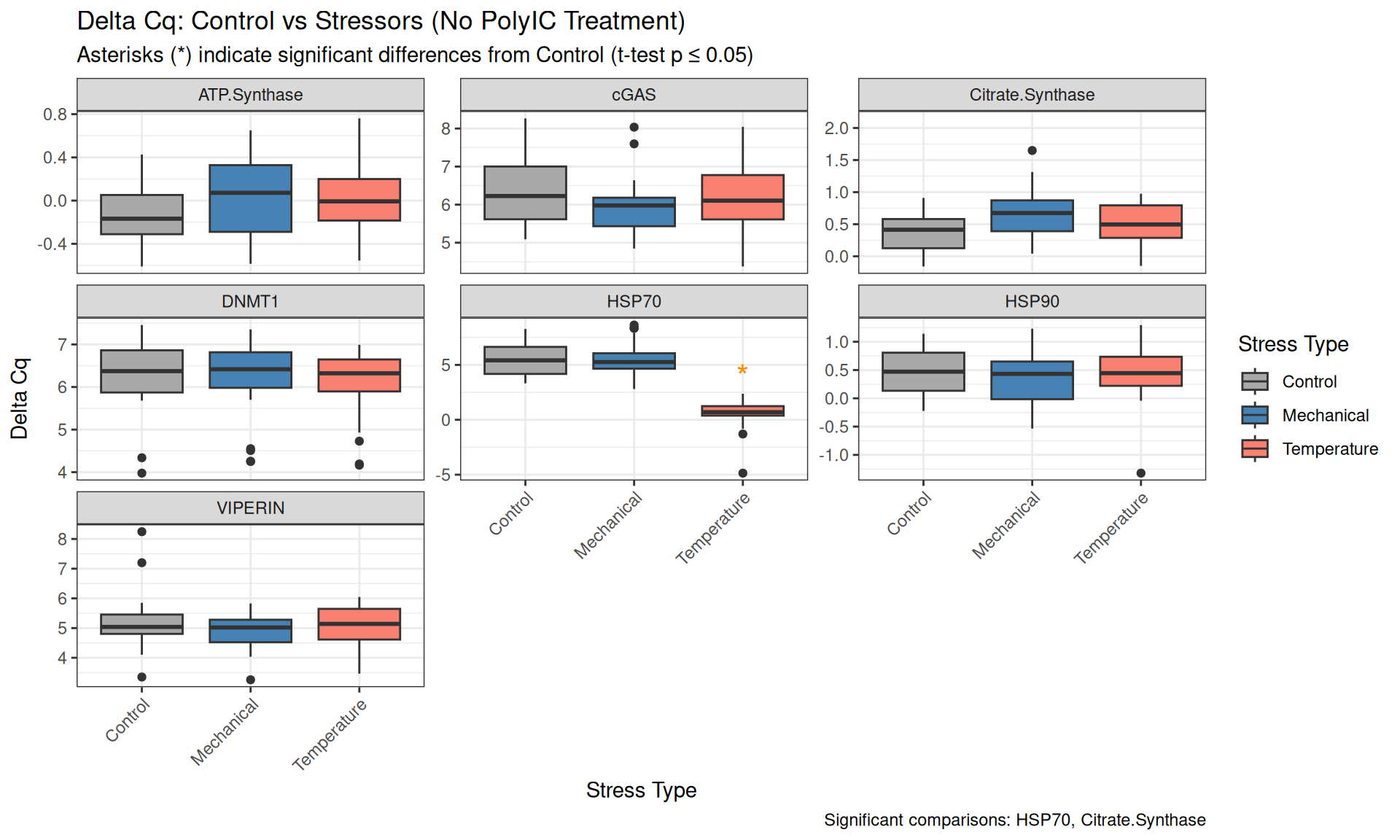
qPCR Analysis - M.gigas PolyIC Data from Valentinas Project
2026
qPCR
Pacific oyster
Magallana gigas
Crassostrea gigas
polyIC
HSP70
HSP90
cGAS
VIPERIN
ATP Synthase
Citrate Synthase
DNMT1
GAPDH
qPCR - M.gigas Valentina PolyIC VIPERIN
2026
Magallana gigas
Crassostrea gigas
Pacific oyster
qPCR
PolyIC
VIPERIN
SsoFast
CFX Connect
CFX96
qPCRs - M.gigas Valentina PolyIC ATP synthase GAPDH HSP70 and HSP90
2026
Magallana gigas
Crassostrea gigas
Pacific oyster
qPCR
PolyIC
ATP synthase
GAPDH
HSP70
HSP90
SsoFast
CFX Connect
CFX96
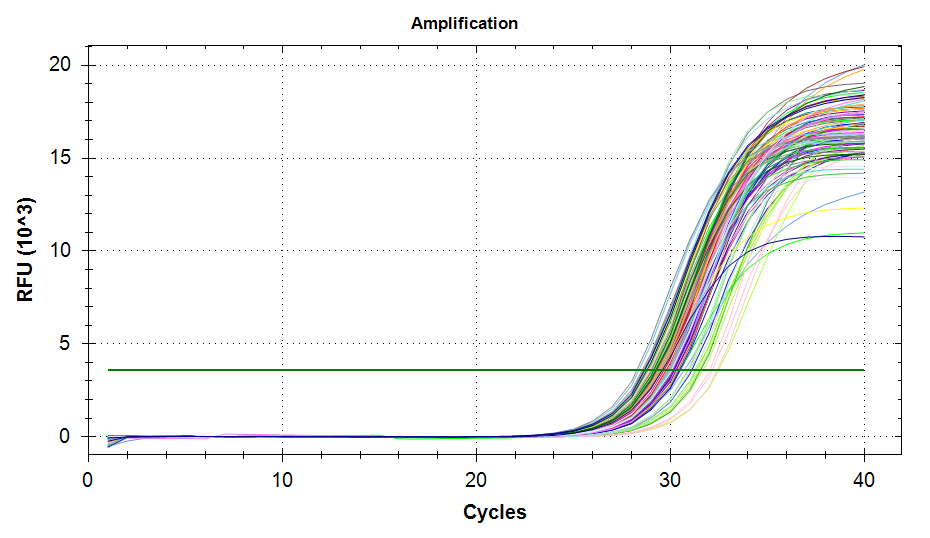
qPCRs - M.gigas Valentina PolyIC cGAS citrate_synthase and DNMT1
2026
Magallana gigas
Crassostrea gigas
Pacific oyster
qPCR
PolyIC
DNMT1
cGAS
citrate_synthase
SsoFast
CFX Connect
CFX96
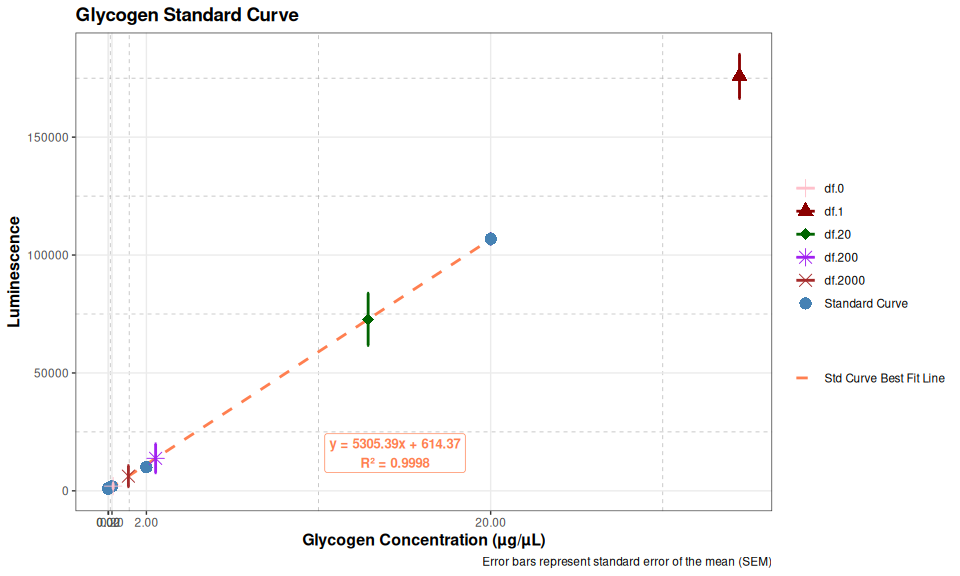
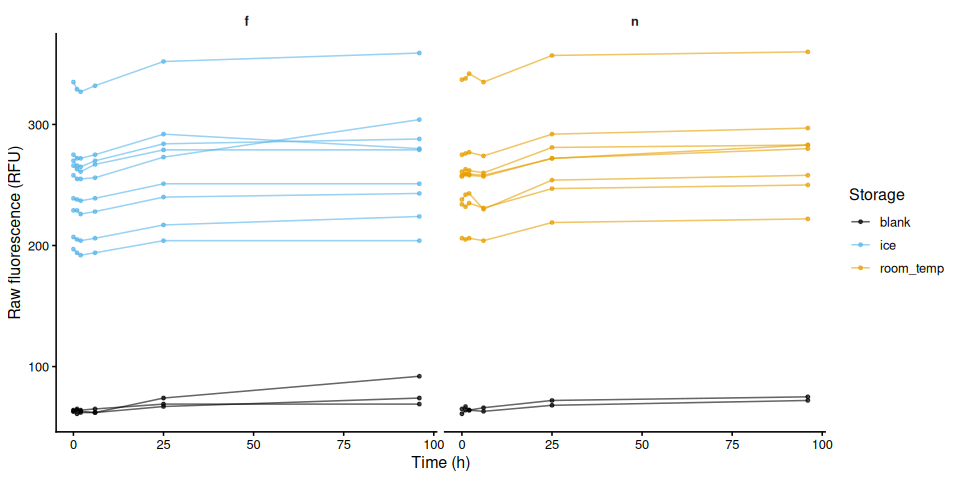
Glycogen Assay - Glycogen-Glo Dilution Testing with M.gigas Ctenidia Homogenate
2026
ctenidia
Glycogen-Glo
glycogen
Pacific oyster
Crassostrea gigas
Magallana gigas
plate reader
Gen 5
Synergy HTX

Data Received - P.tuahiniensis PacBio Genome Sequencing
2025
PacBio
Pocillopora tuahiniensis
sequencing
Data Received
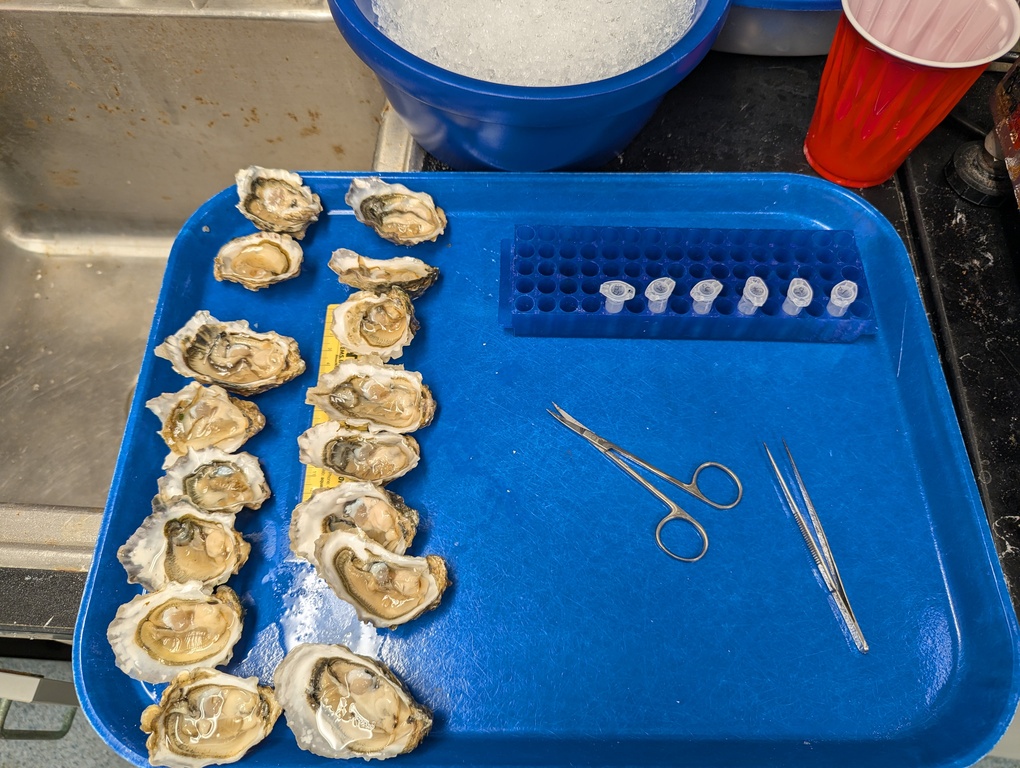
Tissue Collection and Homogenization - M.gigas Ctenidia for Glycogen-Glo Assay
2025
Magallana gigas
Crassostrea gigas
Pacific oyster
ctenidia
Glycogen-Glo
Reverse Transcription - C.gigas PolyIC Mechanical Trials RNA for Valentina from 20251002
Crassostrea gigas
Pacific oyster
Qubit
RNA
Magallana gigas
Poly IC
reverse transcription
Method Testing - E5 Timeseries Multi-species Multi-omics Using workflow-stdm-02
2025
Acropora pulchra
Porites evermanni
Pocillopora tuahiniensis
E5
coral
timeseries_molecular
workflow-stdm-02
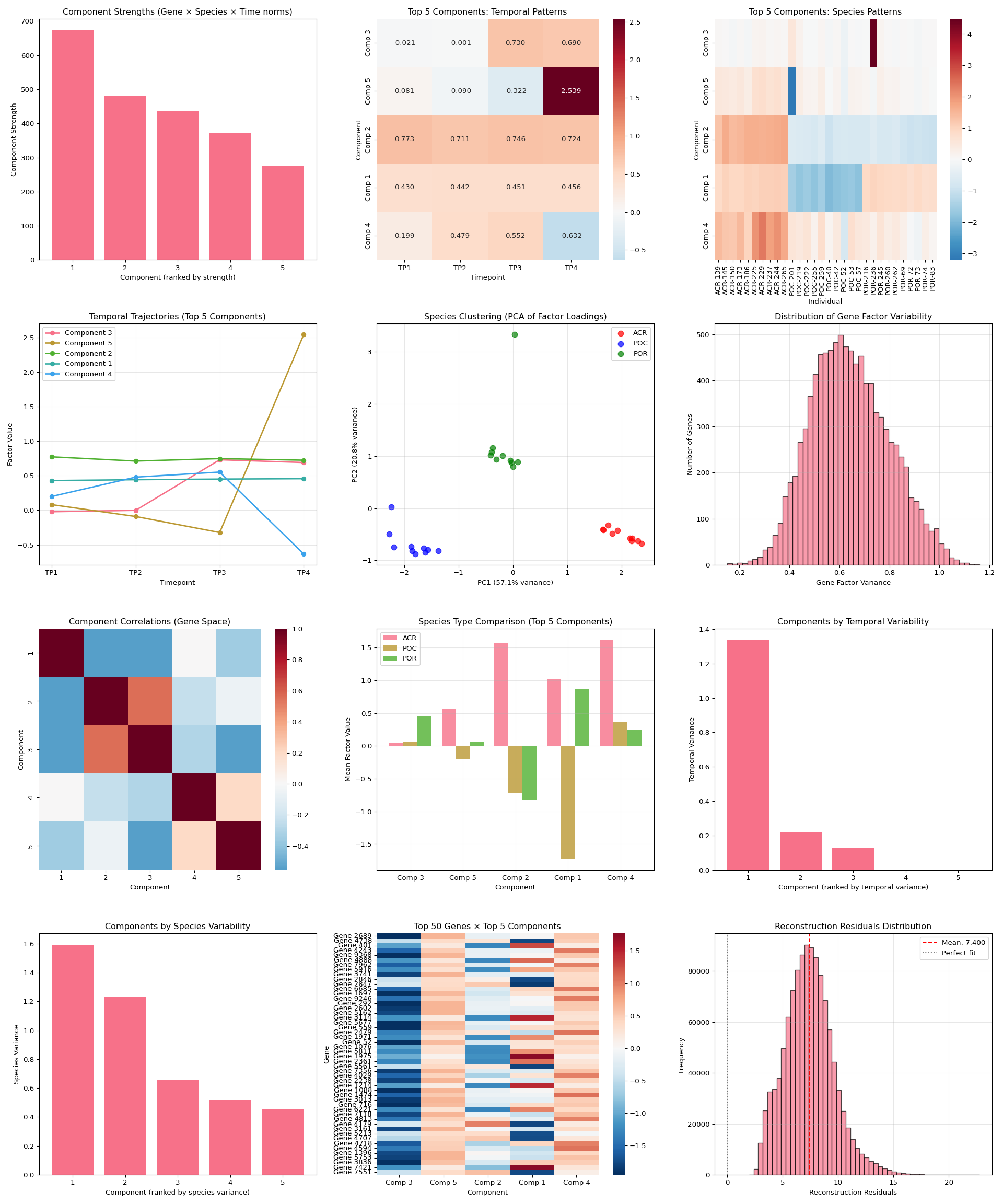
Data Exploration - E5 Timeseries Multi-species Multi-omics Using workflow-stdm
2025
Acropora pulchra
Porites evermanni
Pocillopora tuahiniensis
E5
coral
timeseries_molecular
workflow-stdm
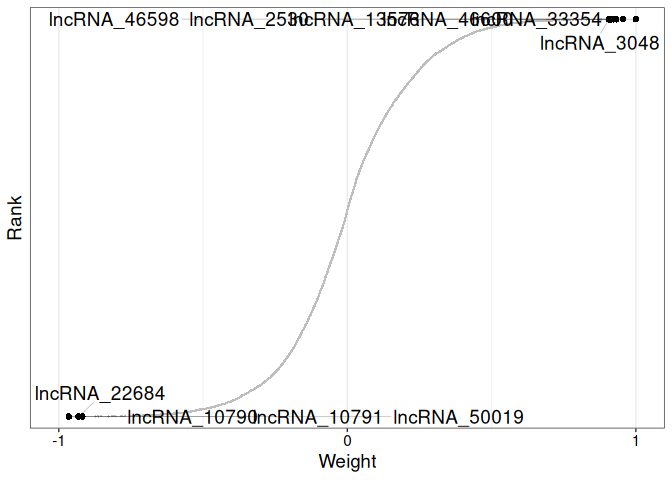
Data Exploration - E5 Timeseries Multi-species Multi-omics Using Barnacle
2025
barnacle
Acropora pulchra
Porites evermanni
Pocillopora tuahiniensis
E5
coral
timeseries_molecular
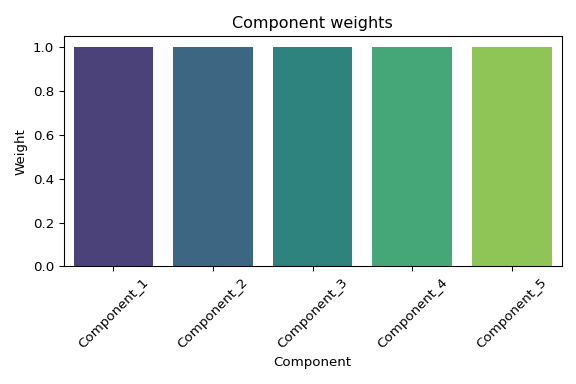
Data Exploration - E5 Timeseries A.pulchra Multi-omics Using MOFA2
2025
MOFA2
Acropora pulchra
E5
coral
timeseries_molecular
RNA Quantification - C.gigas PolyIC Mechanical Trials RNA from 20250902
2025
Crassostrea gigas
Pacific oyster
Qubit
RNA Quantfication
Magallana gigas
Poly IC
RNA Isolation - C.gigas PolyIC Mechanical Trials for Valentina Using Quick-DNA RNA Miniprep Plus Kit
2025
PolyIC
RNA Isolation
Qubit
Magallana gigas
Pacific oyster
Quick DNA/RNA Miniprep Plus Kit
Homogenization - C.gigas Juvenile Whole Body from polyIC Temp and Mech Stress
2025
PolyIC
RNA Isolation
RNA quantification
Qubit
Magallana gigas
Pacific oyster
Data Received - Pacific cod RNA-seq from Psomagen Project AN00025268
2025
AN00025268
RNA-seq
Pacific cod
Gadus macrocephalus
Data Received

RNA Isolation and Quantification - C.gigas PolyIC Mechanical Trials for Valentina Using Quick-DNA RNA MiniPrep Plus Kit
2025
PolyIC
RNA Isolation
RNA quantification
Qubit
Magallana gigas
Pacific oyster
Data Received - Pacific cod Whole Genome Bisulfite Sequencing from Psomagen Project AN00025267
2025
AN00025267
WGBS
Pacific cod
Gadus macrocephalus
Data Received
DNA Isolation Quantification and Concentration - S.namaycush Siscowet Liver Samples Using Zymo Quick-DNA RNA MiniPrep Plus Kit
2025
Salvelinus namaycush
Lake trout
siscowet
DNA isolation
DNA quantification
Qubit
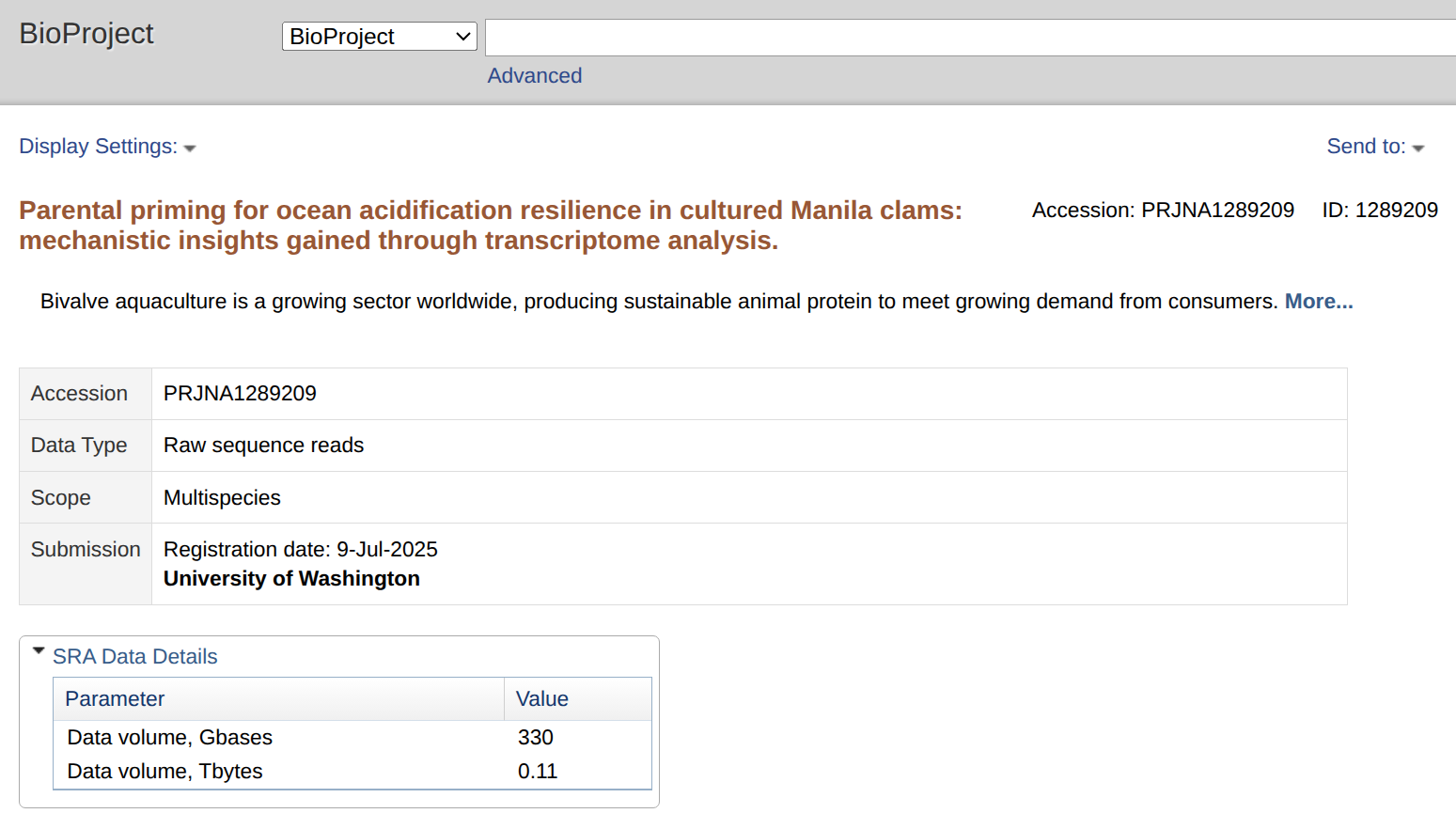
SRA Submission - R.philippinarum Egg RNA-seq Data
2025
Ruditapes philippinarum
Manila clam
RNA-seq
SRA submission
NCBI
PRJNA1289209
Samples Submitted - Pacific cod Blood gDNA and Total RNA to Psomagen
2025
Samples Submitted
Pacific cod
Gadus macrocephalus
Psomagen
AN00025267
AN00025268
Data Received - M.capitata sRNA-seq FastQs
2025
30-1155978938
E5
Montipora capitata
sRNA-seq
Data Received
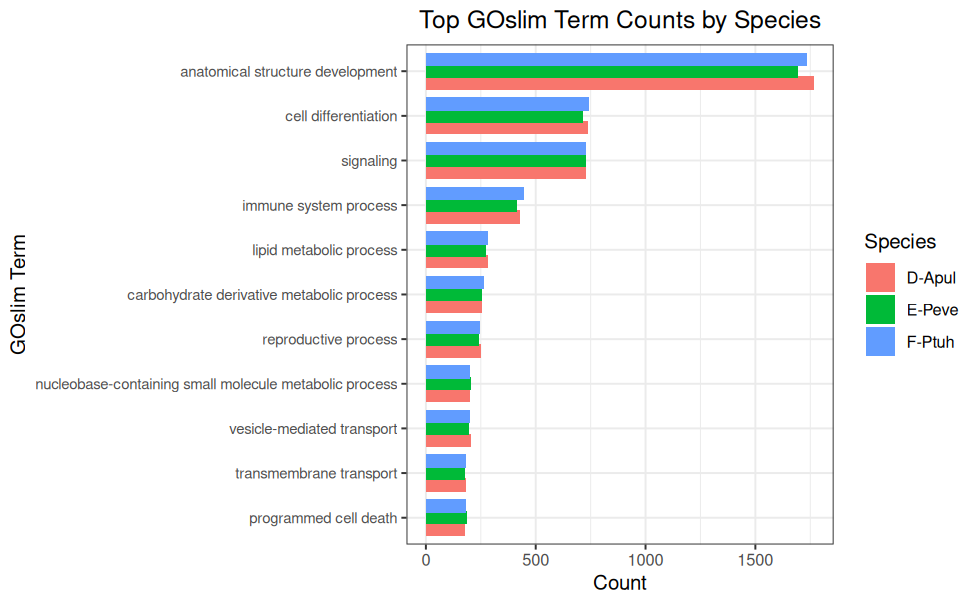
GOslim Counts - P.evermanni Expressed Genes and BLASTing to GOslim for deep-dive-expression Project
2025
E5
deep-dive-expression
GOslim
Porites evermanni
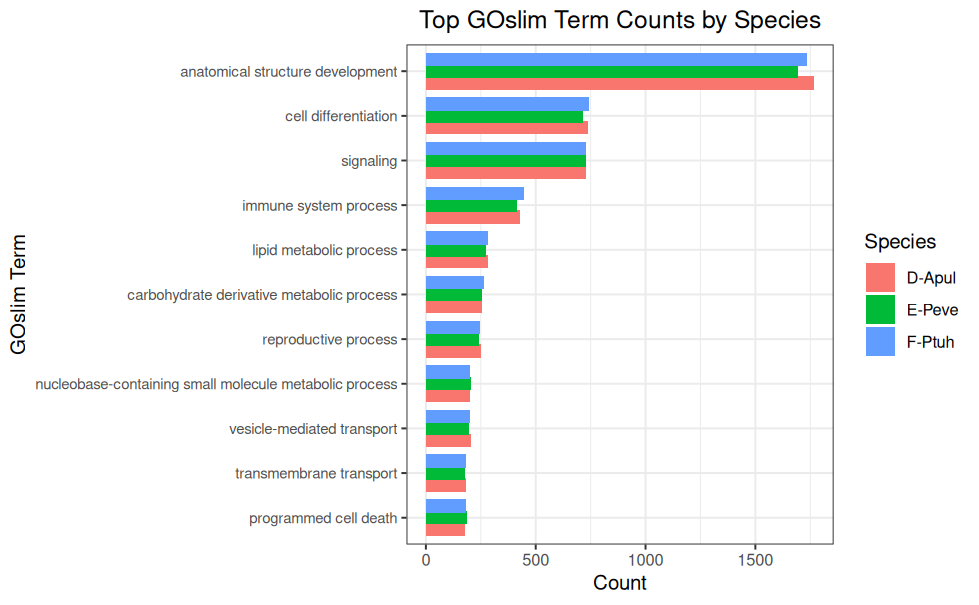
GOslim Counts - P.tuahiniensis Expressed Genes and BLASTing to GOslim for deep-dive-expression Project
2025
E5
deep-dive-expression
GOslim
Pocillopora tuahiniensis
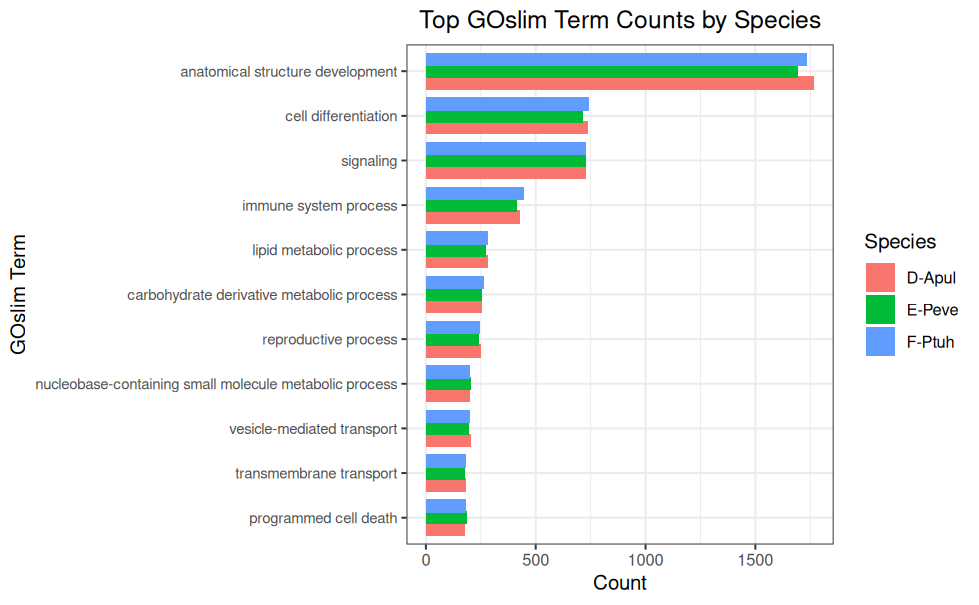
GOslim Counts - A.pulchra Expressed Genes and BLASTing to GOslim for deep-dive-expression Project
2025
Acropora pulchra
E5
RNAseq
deep-dive-expression
GOslim
qPCRs - C.gigas Lifestage Carryover DNMT1 GAPDH HSP70 and HSP90
2025
Crassostrea gigas
Pacific oyster
DNMT1
GAPDH
HSP70
HSP90
SsoFast
CFX Connect
CFX96
project-gigas-carryover
qPCRs - C.gigas Lifestage Carryover ATP_synthase cGAS citrate_synthase and VIPERIN
2025
Crassostrea gigas
Pacific oyster
VIPERIN
citrate synthase
ATP synthase
cGAS
SsoFast
CFX Connect
CFX96
project-gigas-carryover
qPCR - C.gigas Alternative Oxidases Primer Tests
2025
Crassotrea gigas
Pacific oyster
Magallana gigas
primers
AOX
alternative oxidase
XM_066083499.1
XM_034462847.2
NM_001305360.1

Reverse Transcription - C.gigas Lifestage Carryover RNA
2025
reverse transcription
cDNA
GoScript
Crassostrea gigas
Pacific oyster
RNA
project-gigas-carryover
lifestage_carryover
RNA Isolation - C.gigas Lifestage Carryover Samples Using Direct-zol RNA MiniPrep Plus Kit
2025
Crassostrea gigas
Pacific oyster
RNA isolation
Direct-zol RNA MiniPrep Plus Kit
Magallana gigas
RNA Quantification - C.gigas Lifestage Carryover RNA Samples Using Qubit High Sensitivity RNA Assay
2025
Crassostrea gigas
Pacific oyster
RNA quantification
Qubit
Magallana gigas
RNA Isolation - C.gigas Lifestage Carryover Samples Using Direct-zol RNA MiniPrep Plus Kit
2025
Crassostrea gigas
Pacific oyster
RNA isolation
Direct-zol RNA MiniPrep Plus Kit
Magallana gigas
Sample Homogenization - C.gigas Lifestage Carryover Samples Using TriZol and Bullet Blender
2025
Bullet Blender
homogenization
TriZol
Crassostrea gigas
Pacific oyster
Magallana gigas
DNA RNA Isolation and Quantification - G.macrocephalus Blood Using Bullet Blender and Zymo Quick DNA RNA MiniPrep
2025
Bullet Blender
Pacific cod
Gadus macrocephalus
blood
DNA isolation
RNA isolation
DNA quantification
RNA quantification
Qubit
DNA RNA Isolation and Quantification - G.macrocephalus Blood Using Bullet Blender and Zymo Quick DNA RNA MiniPrep
2025
Bullet Blender
Pacific cod
Gadus macrocephalus
blood
DNA isolation
RNA isolation
DNA quantification
RNA quantification
Qubit
DNA RNA Isolation and Quantification - G.macrocephalus Blood Using Bullet Blender and Zymo Quick DNA RNA MiniPrep Plus Kit
2025
Bullet Blender
Pacific cod
Gadus macrocephalus
blood
DNA isolation
RNA isolation
DNA quantification
RNA quantification
Qubit
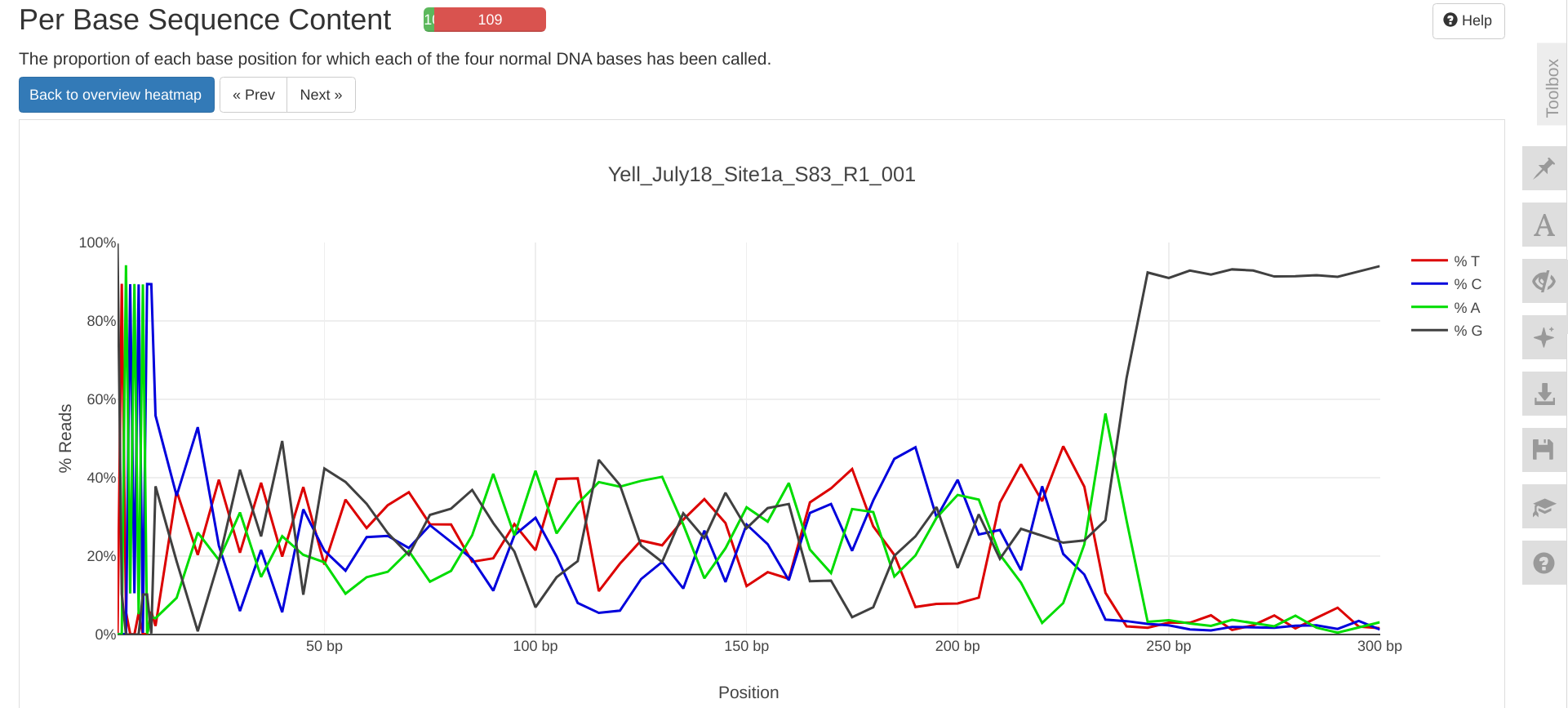
FastQC MultiQC - Yellow Island eDNA Project
2025
eDNA
Yellow Island
FastQC
MultiQC
project-eDNA-yellow
Trimming - Yellow Island eDNA FastQs Using Fastp FastQC and MultiQC
2025
eDNA
Yellow Island
fastp
trimming
FastQC
MultiQC
project-eDNA-yellow

DNA RNA Isolation and Quantification - G.macrocephalus Blood Using Bullet Blender and Zymo Quick DNA RNA MiniPrep Plus Kit
2025
Bullet Blender
Pacific cod
Gadus macrocephalus
blood
DNA isolation
RNA isolation
DNA quantification
RNA quantification
Qubit
DNA RNA Isolation and Quantification - G.macrocephalus Blood Using Bullet Blender and Zymo Quick DNA RNA MiniPrep Plus Kit
2025
Bullet Blender
Pacific cod
Gadus macrocephalus
blood
DNA isolation
RNA isolation
DNA quantification
RNA quantification
Qubit
Samples Submitted - S.namaycush Liver gDNA to Angie Schmoldt at UW-Milwaukee
2025
Samples Submitted
lake trout
gDNA
liver
Salvelinus namaycush

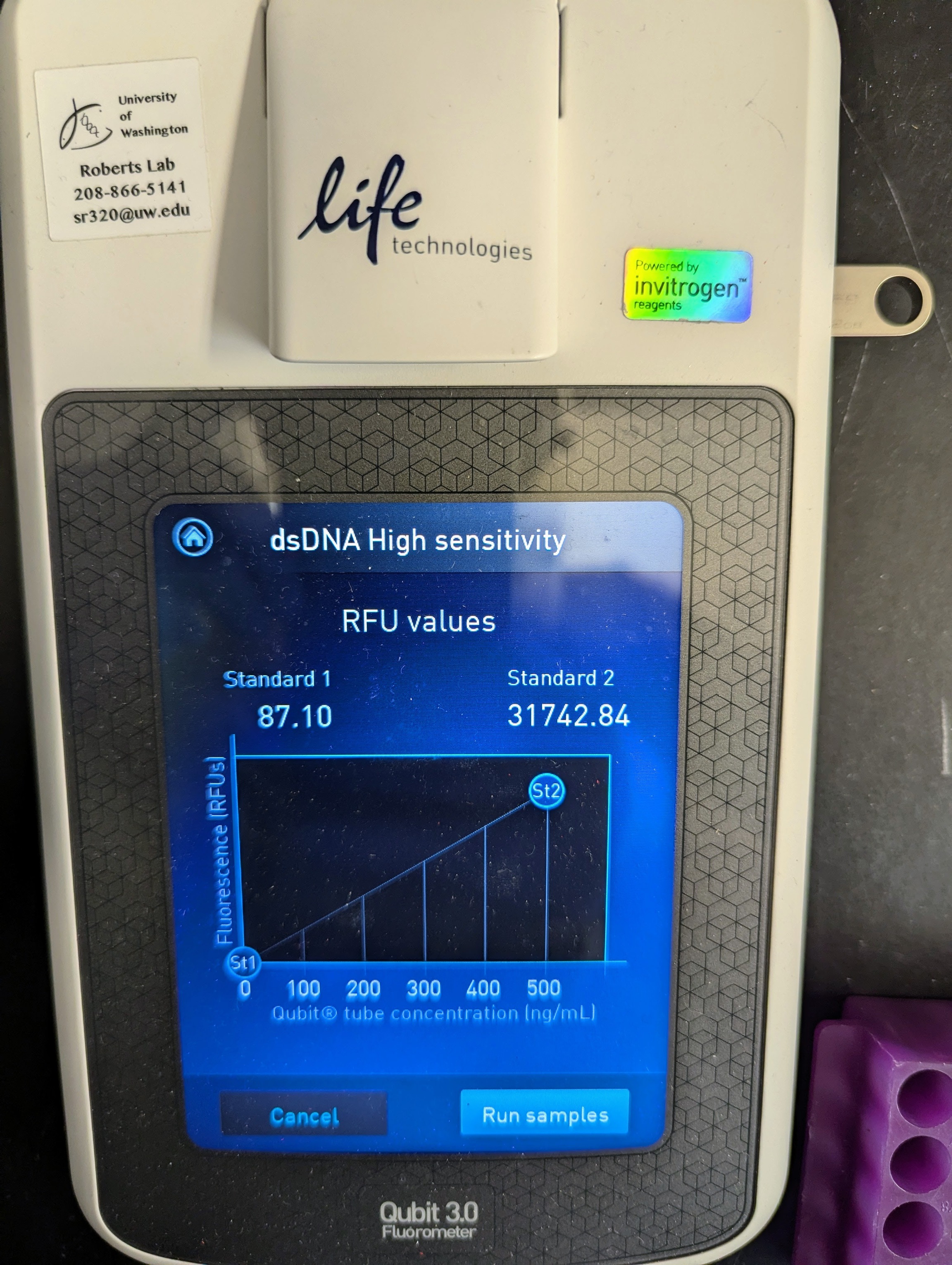
DNA and RNA Isolations and Quantification - G.macrocephalus Blood Using Zymo Quick-RNA RNA MiniPrep Plus Kit
2025
Gadus macrocephalus
Pacific cod
blood
DNA isolation
RNA isolation
Qubit
DNA quantification
RNA quantification

Alignment - P.evermanni RNA-seq Data from E5 timeseries_molecular Project Using Hisat2
2025
Porites evermanni
E5
coral
Hisat2
alignment
RNAseq
Stringtie
Alignment - P.tuahiniensis RNA-seq Data from E5 timeseries_molecular Project Using Hisat2
2025
Pocillopora tuahiniensis
E5
coral
Hisat2
alignment
RNAseq
Stringtie
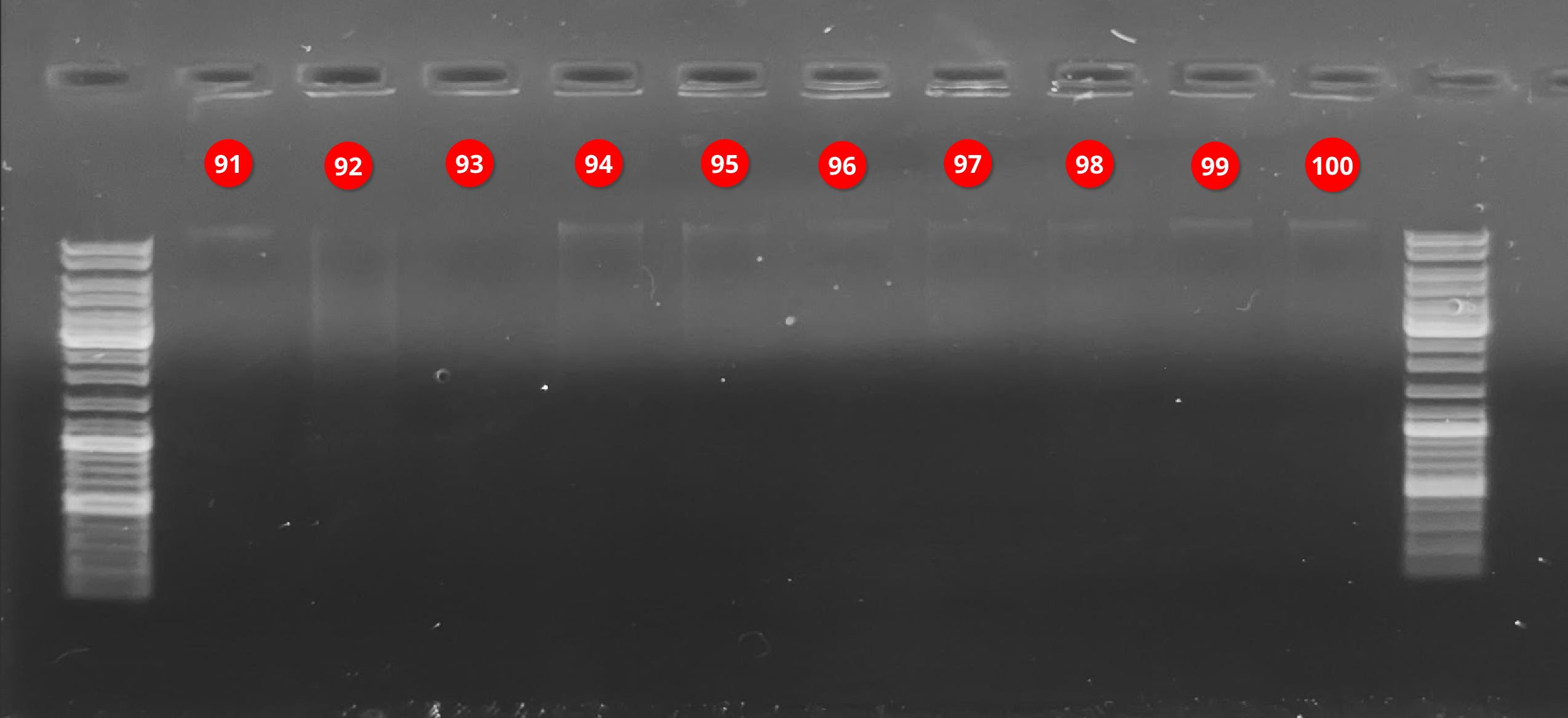
Agarose Gel - S.namaycush Liver gDNA from 20250220
2025
agarose gel
gDNA
liver
Lake trout
Salvelinus namaycush
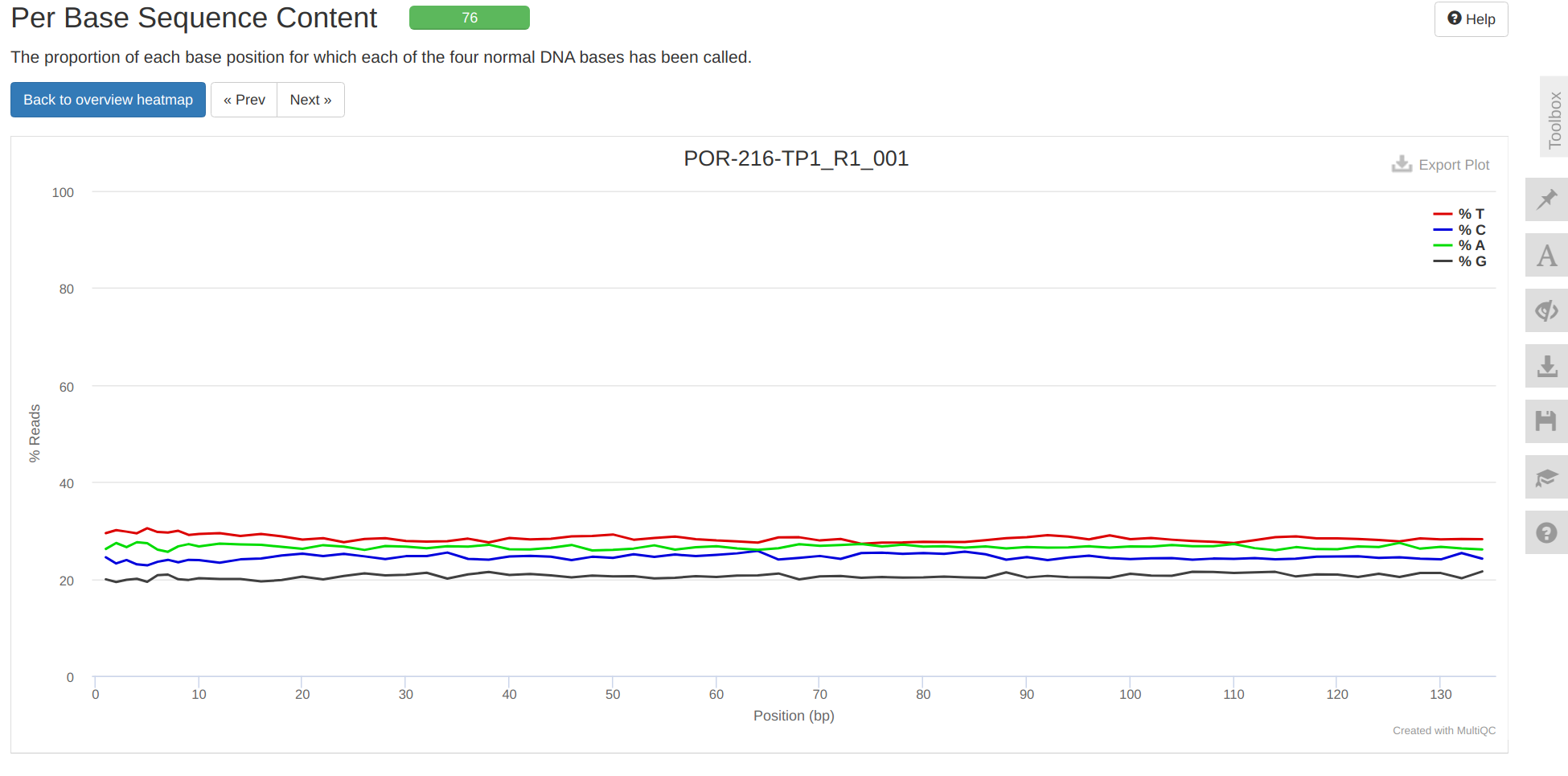
Trimming - P.evermanni RNA-seq Trimming and QC with fastp FastQC and MultiQC for E5 timeseries_molecular Project
2025
Porites evermanni
E5
trimming
fastp
FastQC
MultiQC
RNAseq
timeseries_molecular
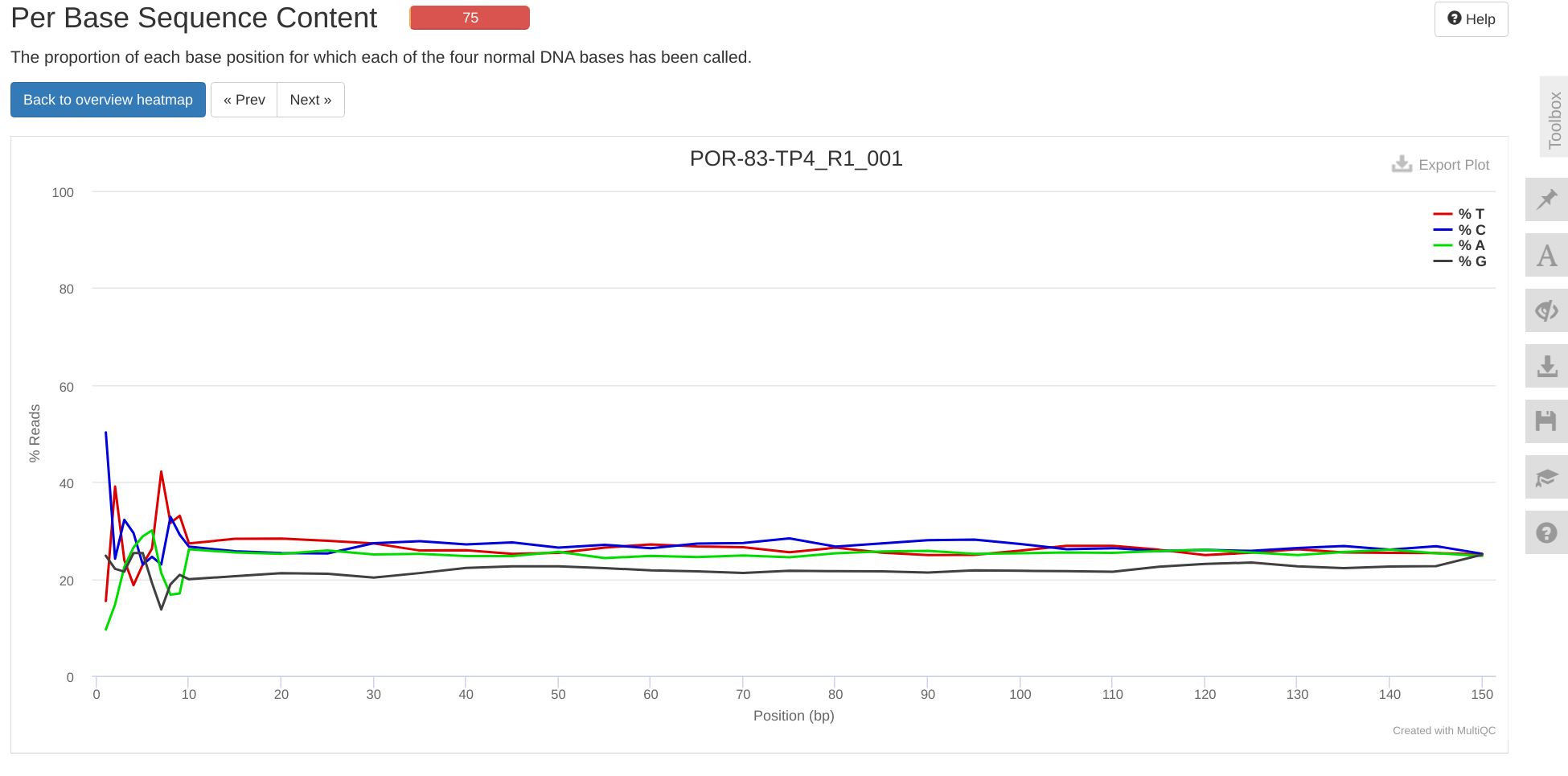
Trimming - P.tuahiniensis RNA-seq Trimming and QC with fastp FastQC and MultiQC for E5 timeseries_molecular Project
2025
Pocillopora tuahiniensis
E5
trimming
fastp
FastQC
MultiQC
RNAseq
timeseries_molecular
DNA Isolation and Quantification - S.namaycush Liver
2025
lake trout
Salvelinus namaycush
DNA isolation
Qubit
DNA quantification
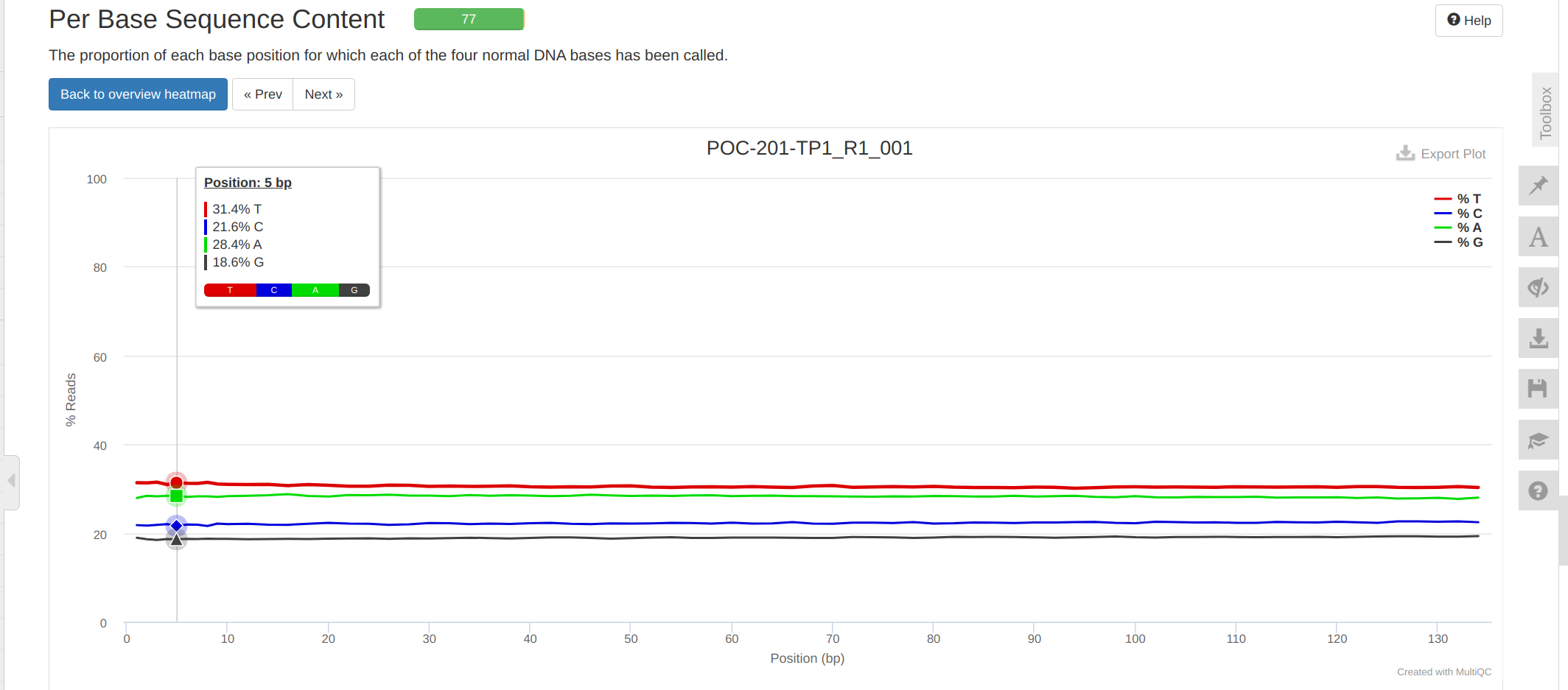
FastQC MultiQC - P.evermanni RNA-seq Data for timeseries_molecular Project
2025
Porites evermanni
E5
FastQC
MultiQC
RNAseq
timeseries_molecular
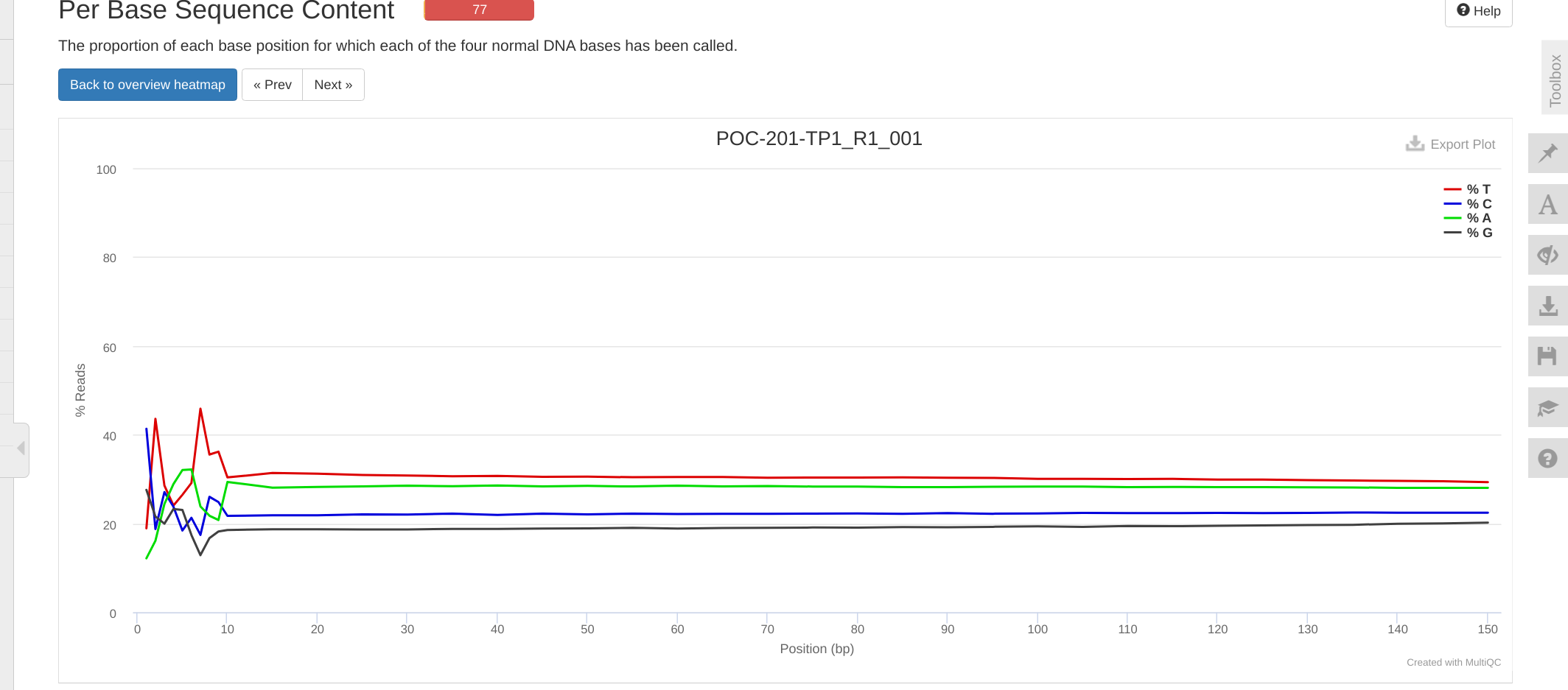
FastQC MultiQC - P.tuahiniensis RNA-seq Data for timeseries_molecular Project
2025
Pocillopora tuahiniensis
E5
FastQC
MultiQC
timeseries_molecular
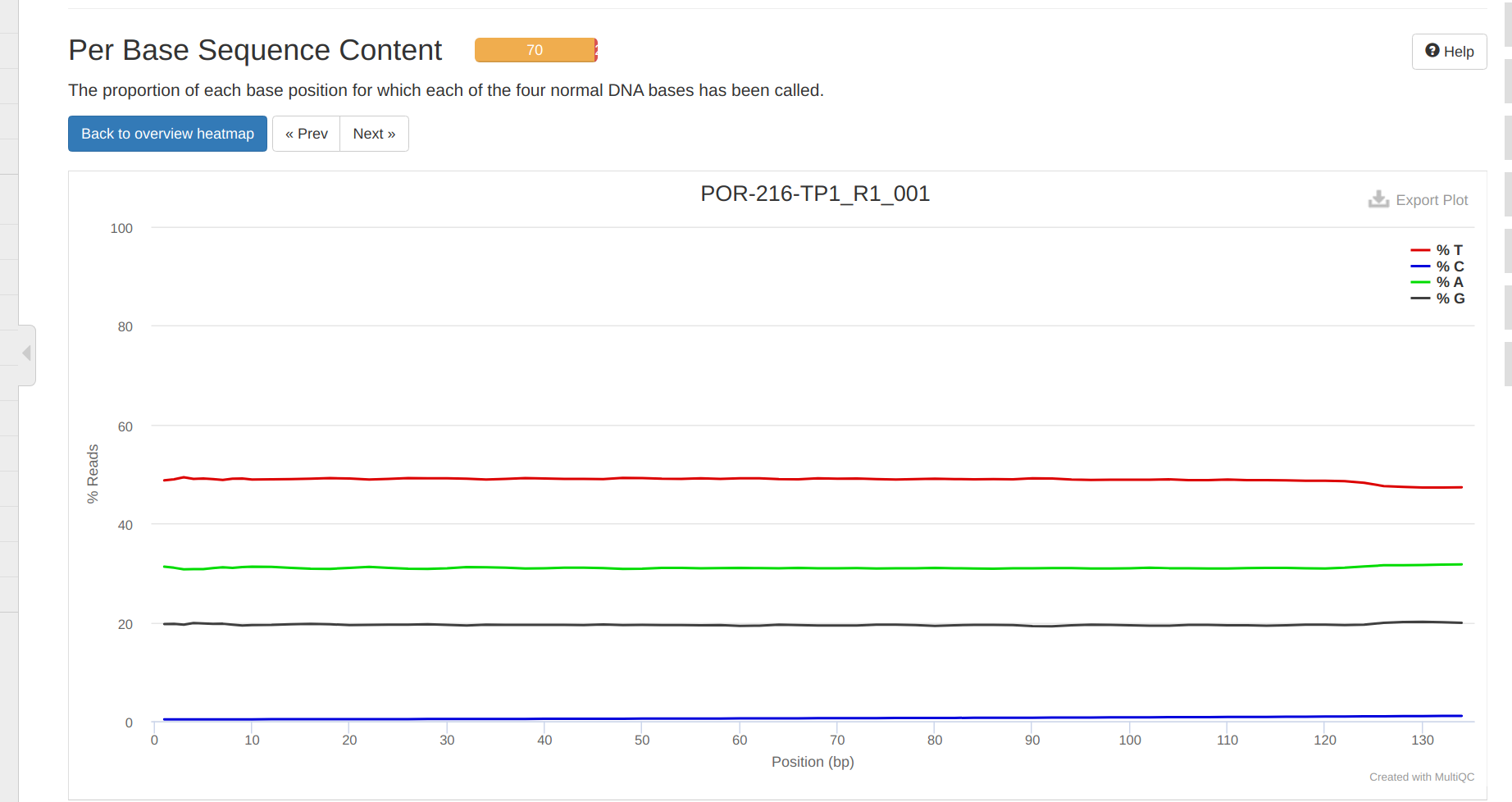
Trimming - P.evermanni WGBS Trimming and QC with fastp FastQC and MultiQC for E5 timeseries_molecular Project
2025
Porites evermanni
E5
WGBS
bisulfite sequencing
FastQC
MultiQC
fastp
trimming
timeseries_molecular
coral
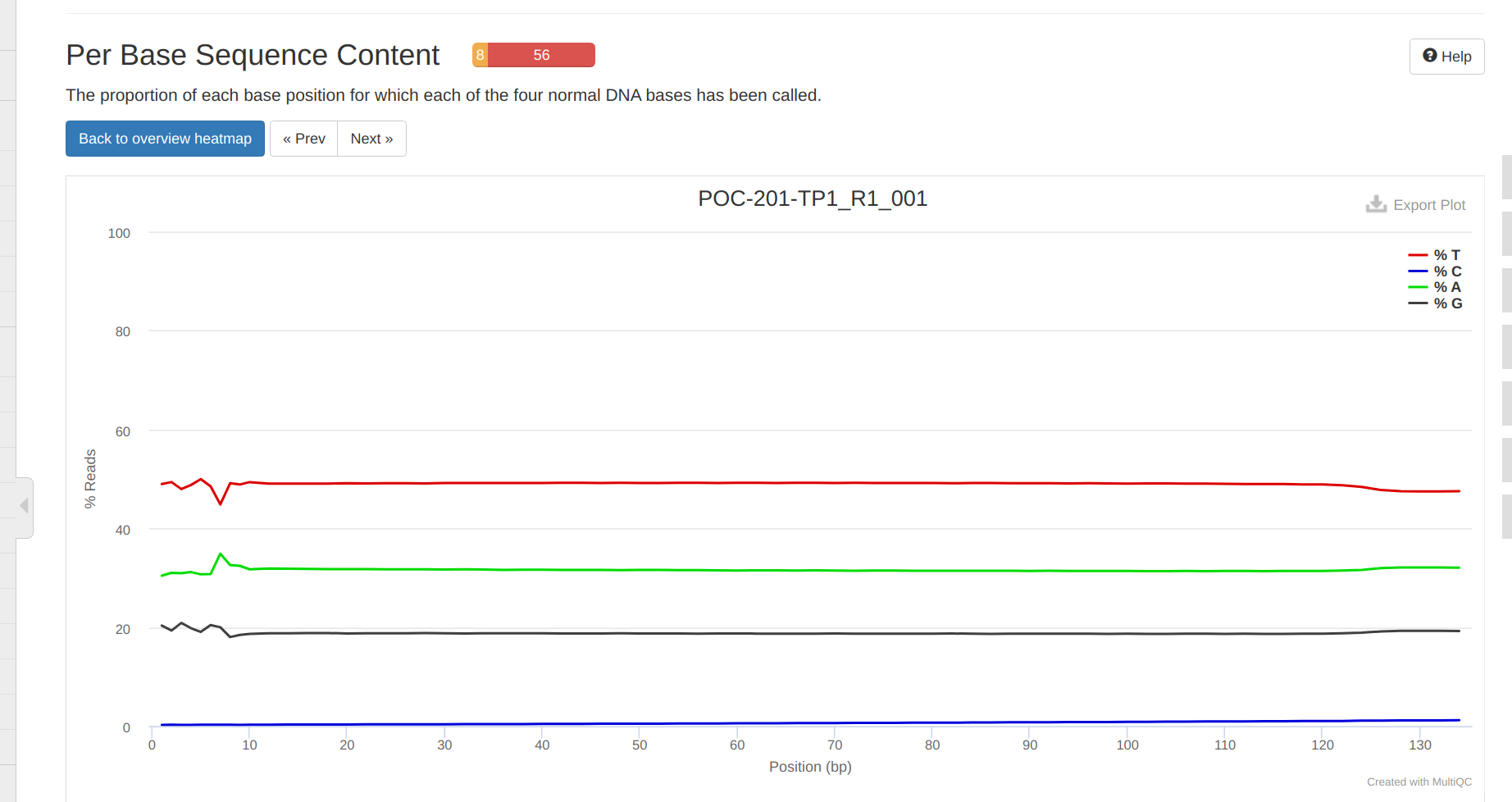
Trimming - P.tuahiniensis WGBS Trimming and QC with fastp FastQC and MultiQC for E5 timeseries_molecular Project
2025
Porites tuahiniensis
E5
WGBS
bisulfite sequencing
FastQC
MultiQC
fastp
trimming
timeseries_molecular
coral
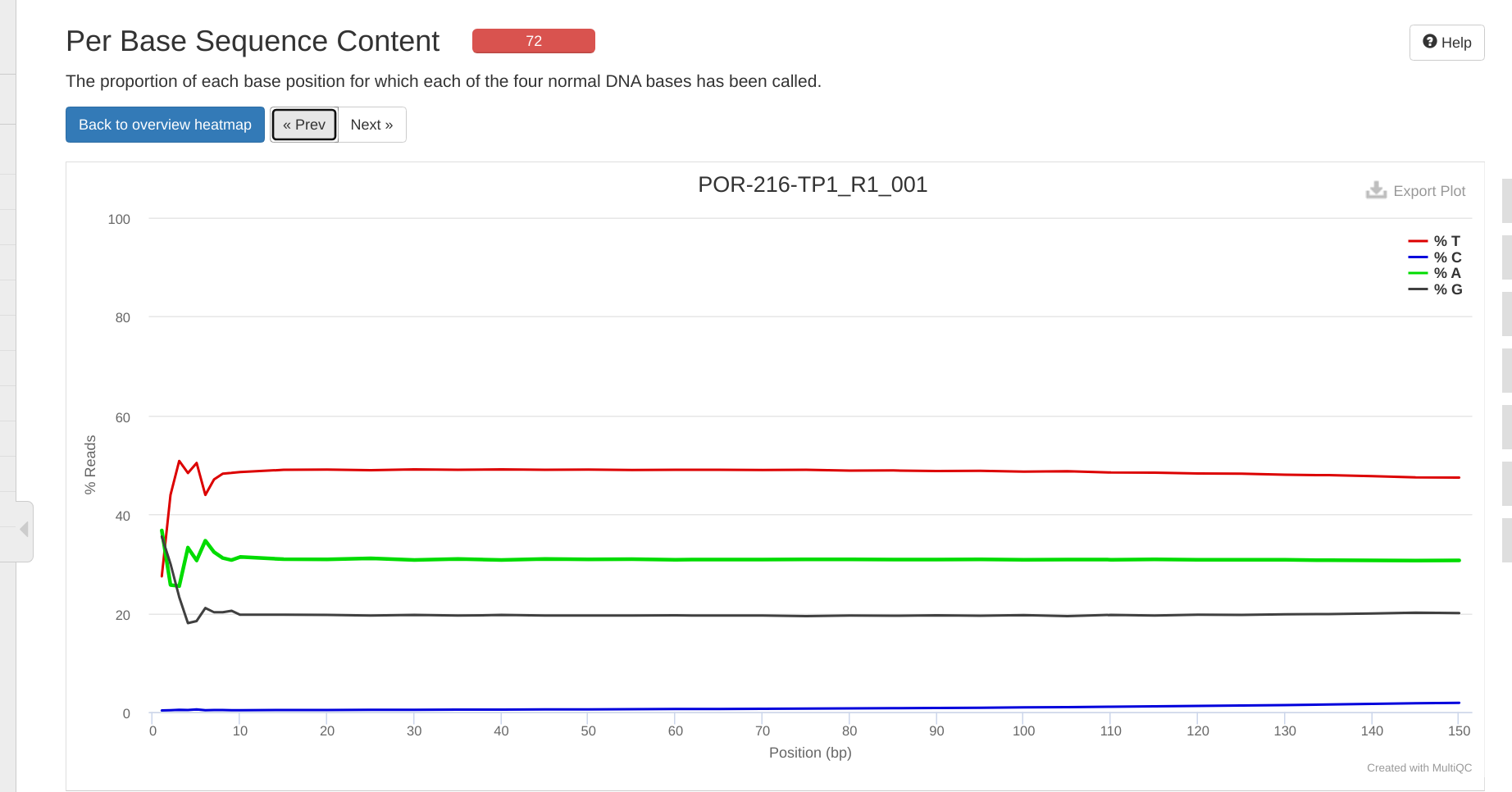
FastQC MultiQC - P.evermanni Raw WGBS Renaming and FastQC MultiQC E5 timeseries_molecular Project
2025
Porites evermanni
E5
WGBS
bisulfite sequencing
FastQC
MultiQC
timeseries_molecular
coral
FastQC MultiQC - P.tuahiniensis Raw WGBS Renaming and FastQC MultiQC timeseries_molecular Project
2025
Pocillopora tuahiniensis
E5
WGBS
bisulfite sequencing
FastQC
MultiQC
timeseries_molecular
coral
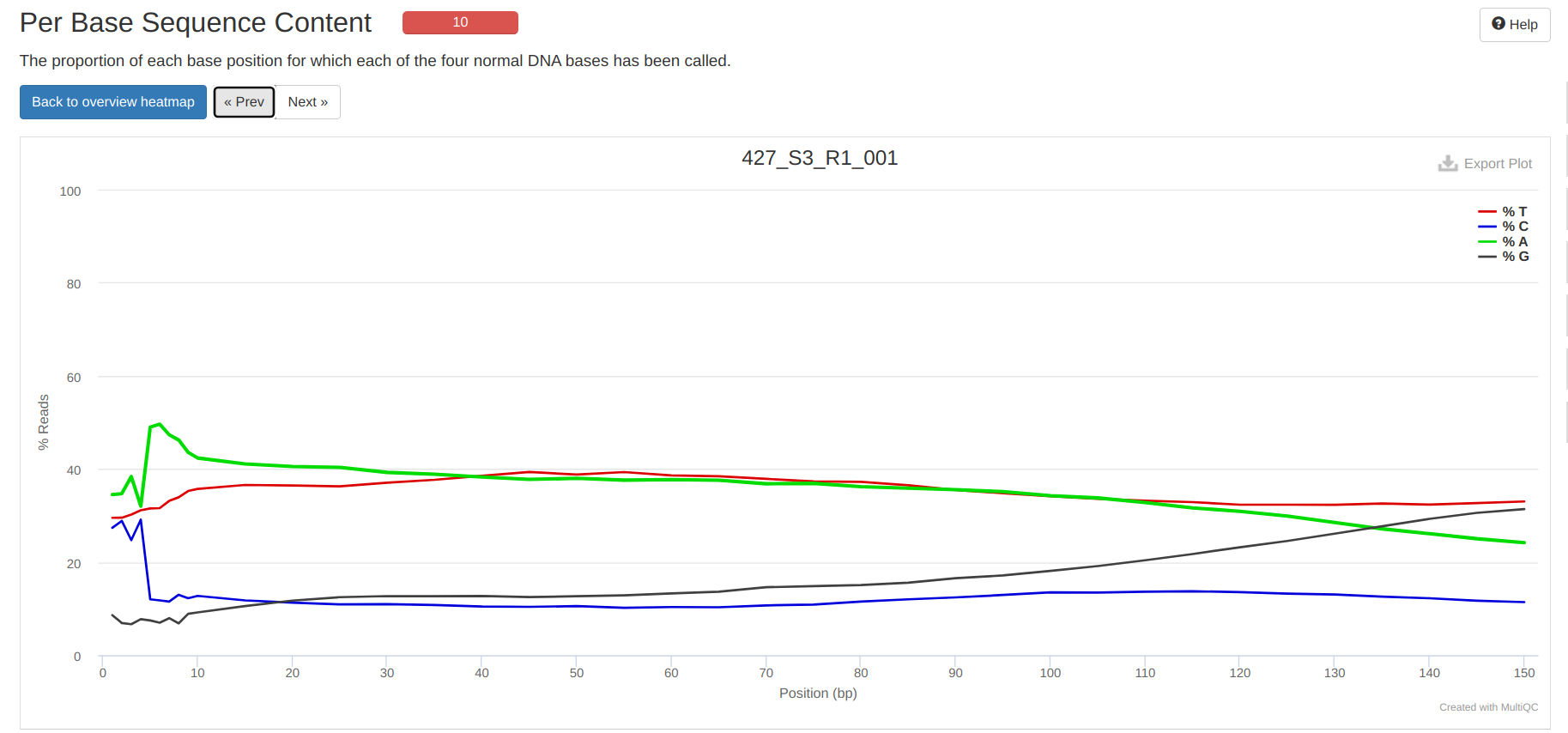
FastQC MultiQC - A.pulchra WGBS E5 Deep Dive Expression
2025
Acropora pulchra
E5
deep-dive-expression
FastQC
MultiQC
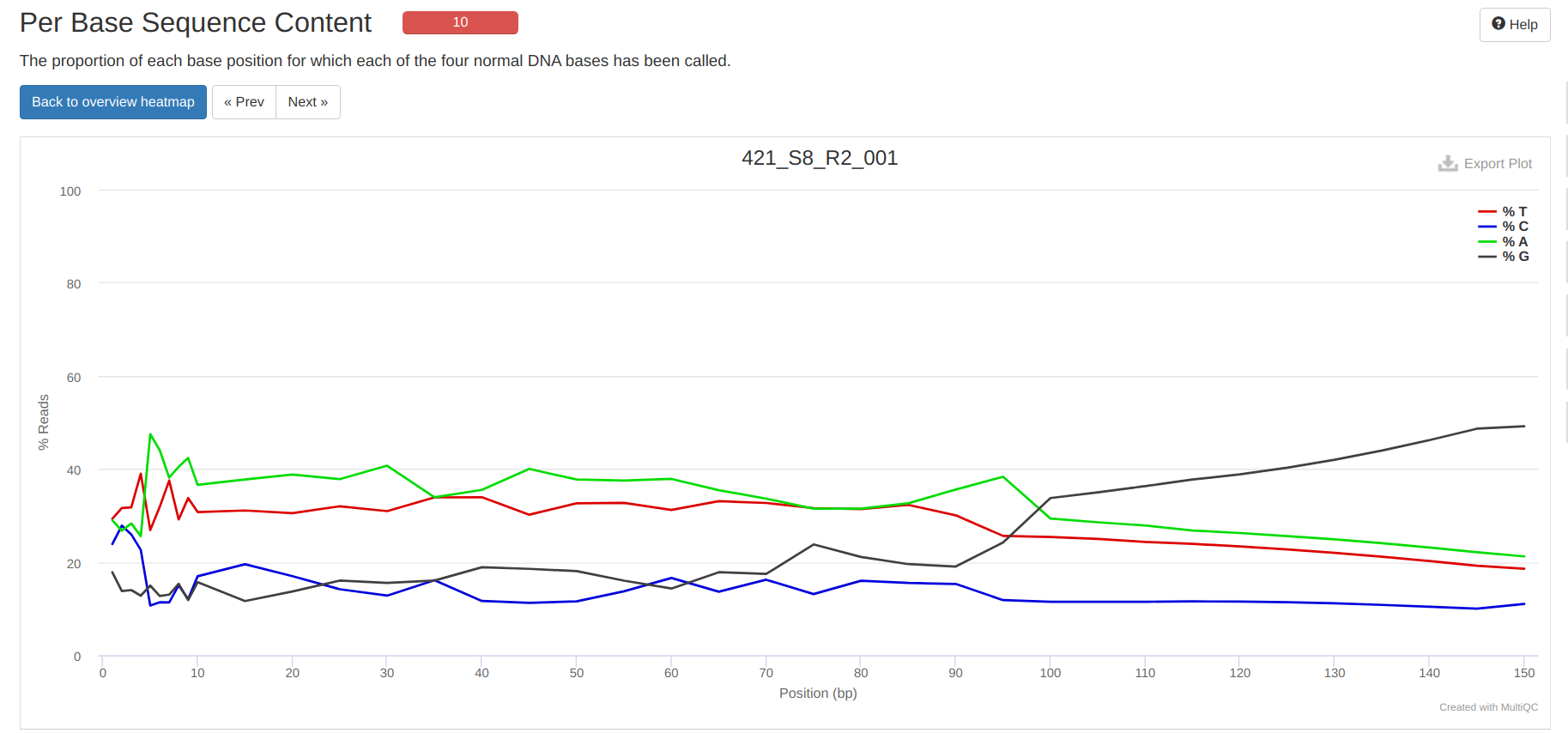
FastQC MultiQC - P.evermanni WGBS E5 Deep Dive Expression
2025
Porites evermanni
E5
deep-dive-expression
FastQC
MultiQC
WGBS
BS-seq
bisulfite sequencing
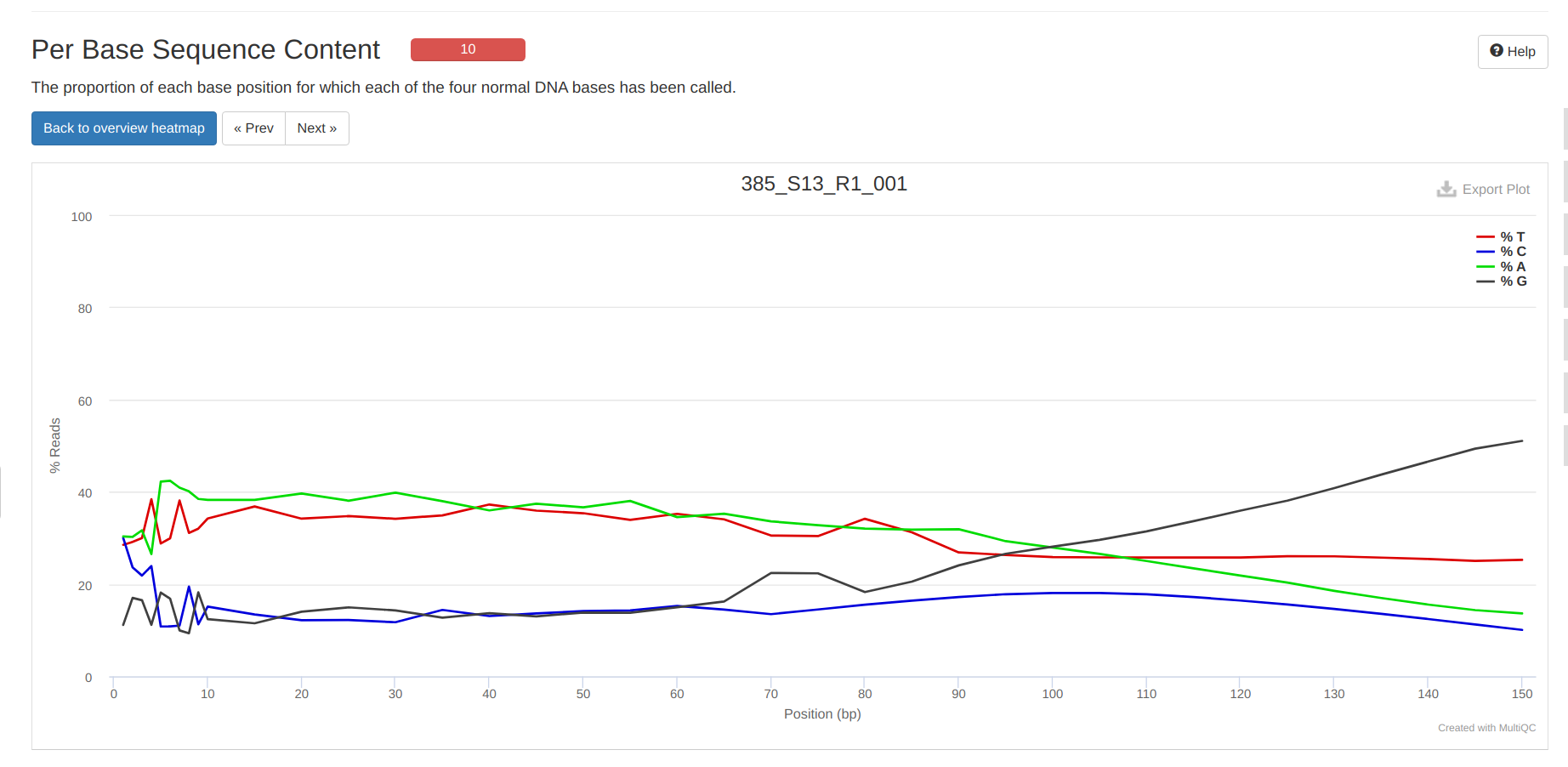
FastQC MultiQC - P.tuahiniensis WGBS E5 Deep Dive Expression
2025
Pocillopora tuahiniensis
E5
deep-dive-expression
FastQC
MultiQC
WGBS
BS-seq
bisulfite sequencing
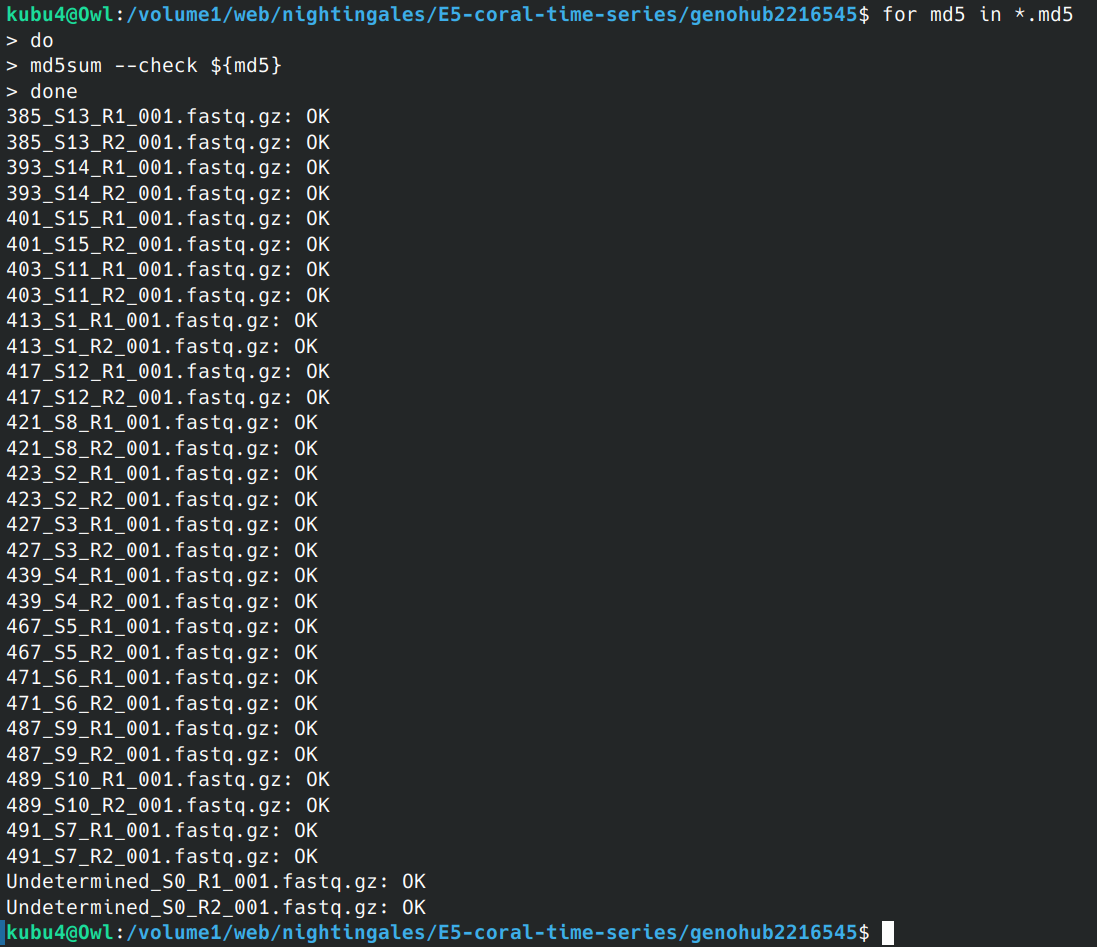
Data Received - E5 Coral WGBS from Genohub Project 2216545 Using Rclone on Owl
2025
E5
coral
WGBS
BS-seq
bisulfite sequencing
Genohub
2216545
Data Received
rclone
Differential Expression - R.philippinarum miRNAs Using DEseq2
2025
Ruditapes philippinarum
Manila clam
DEseq2
sRNA-seq
miRNA
Computer Management - Fix Most of Raven Boot Issues
2025
raven
Computer Management
Bisulfite Genome - P.evermanni BS Genome Using Bismark
2025
Porites evermanni
E5
timeseries_molecular
bismark
bisulfite genome
Bisulfite Genome - P.tuahiniensis BS Genome Using Bismark
2025
Pocillopora tuahiniensis
bismark
bisulfite
coral
E5
timeseries_molecular
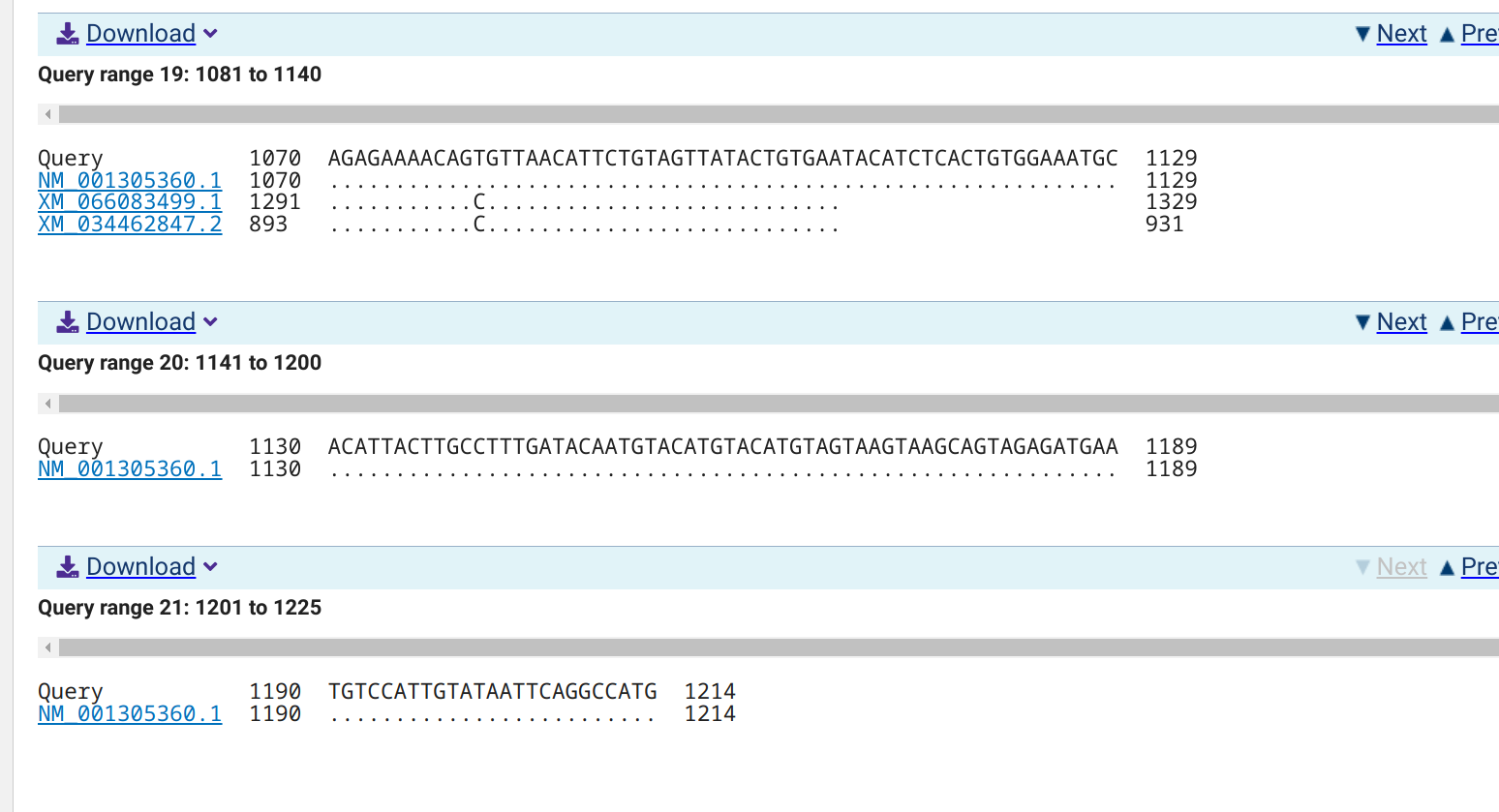
Primer Design - C.gigas Alternative Oxidase AOX Variants Using Primer3
2025
Crassotrea gigas
Pacific oyster
Primer3
EMBOSS
PrimerSearch
Magallana gigas
primers
AOX
alternative oxidase
Data Wrangling - Reformat P.tuahiniensis GFF to Valid GFF3 Using awk genometools and AGAT
2025
GFF
genometools
E5
timeseries_molecular
Porites tuahiniensis
coral
AGAT
Data Wrangling - Reformat P.evermanni GFF to Valid GFF3 Using awk genometools and AGAT
2025
GFF
genometools
E5
timeseries_molecular
Porites evermanni
coral
AGAT
Bismark Alignments - A.pulchra Trimmed Reads Using Bismark on Hyak
2025
Bismark
Acropora pulchra
coral
timeseries_molecular
E5
klone
hyak
alignment
WGBS
Bisulfite Genome - A.pulchra BS Genome Using Bismark
2025
Acropora pulchra
bismark
bisulfite
coral
E5
timeseries_molecular
Trimming - A.pulchra WGBS with fastp FastQC and MultiQC on Raven
2025
Acopora pulchra
WGBS
trimming
fastp
FastQC
MultiQC
Raven
FastQC MultiQC - A.pulchra WGBS E5 Timeseries
2024
E5
timeseries_molecular
WGBS
Acropora pulchra
FastQC
MultiQC
Data Received - Coral E5 WGBS Time Series from Azenta Project 30-1067895835
2024
E5
30-1067895835
WGBS
BS-seq
bisulfite
Data Received
timeseries_molecular
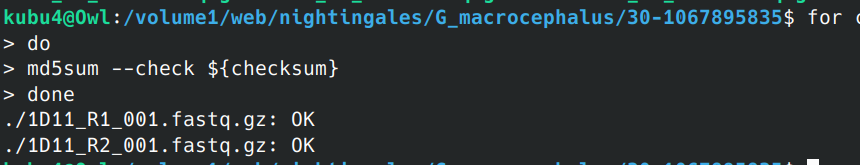
Data Received - G.macrocephalus WGBS Data from Azenta Project 30-1067895835
2024
WGBS
BS-seq
Data Received
bisulfite
30-1067895835
Pacific cod
Gadus macrocephalus
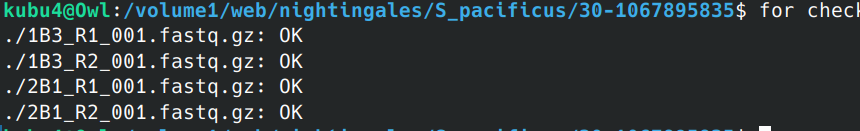
Data Received - S.pacificus WGBS Data from Azenta Project 30-1067895835
2024
WGBS
BS-seq
Data Received
bisulfite
30-1067895835
Somniosus pacificus
sleeper shark

qPCR Analysis - C.gigas Lifestages Carryover
2024
Crassostrea gigas
Pacific oyster
qPCR
project-gigas-carryover
lifestage_carryover
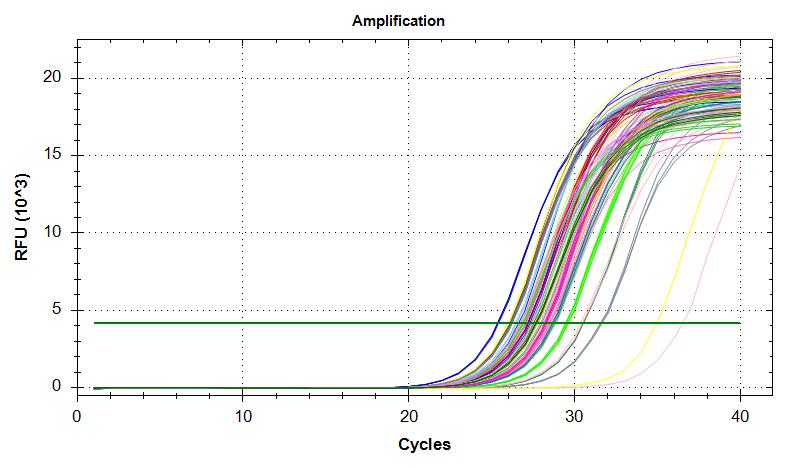
qPCR - C.gigas Lifestage Carryover VIPERIN
2024
Crassostrea gigas
Pacific oyster
SsoFast
CFX Connect
CFX96
project-gigas-carryover
VIPERIN
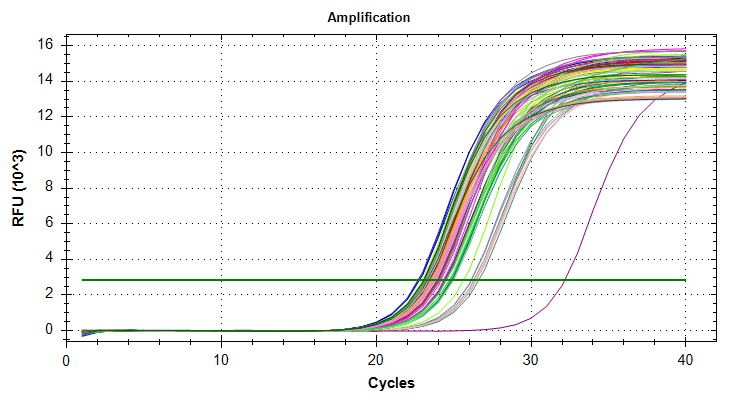
qPCRs - C.gigas Lifestage Carryover citrate synthase DNMT1 HSP70 and HSP90
2024
Crassostrea gigas
Pacific oyster
SsoFast
CFX Connect
CFX96
project-gigas-carryover
citrate synthase
DNMT1
HSP70
HSP90
Data Received - Coral E5 sRNA-seq Time Series Azenta Project 30-1069297013
2024
coral
E5
sRNA-seq
timeseries_molecular
Data Received
30-1069297013
miRNA Identification - R.phillipinarum sRNA-seq Using ShortStack and miRBase Mature miRNAs
2024
Ruditapes phillipinarum
Manila clam
sRNA-seq
miRNA
project-clam-oa
ShortStack
miRBase

qPCRs - C.gigas Lifestage Carryover GAPDH ATP_synthase and cGAS
2024
Crassostrea gigas
Pacific oyster
GAPDH
ATP synthase
cGAS
SsoFast
CFX Connect
CFX96
project-gigas-carryover

Trimming and Merging - R.philippinarum sRNA-seq Data Using fastp FastQC and MultiQC
2024
Ruditapes philippinarum
Manila clam
project-clam-oa
sRNA-seq

FastQ Concatentation and QC - R.philippinarum sRNA-seq Data Using FastQC and MultiQC
2024
Manila clam
Ruditapes philippinarum
30-1035633055
30-1035633055-TS01
project-clam-oa
FastQC
MultiQC

Methylation Extraction - C.virginica CEASMALLR WGBS Using Bismark
2024
Crassostrea virginica
Eastern oyster
ceasmallr
wgbs
BS-seq
Bismark
methylation extraction

Deduplication - C.virginica CEASMALLR WGBS Using Bismark
2024
Crassostrea virginica
Eastern oyster
ceasmallr
Bismark
WGBS
BS-seq
deduplication
Alignment - C.virginica CEASMALLR WGBS Using Bismark on Klone Array
2024
trimming
Crassostrea virginica
Eastern oyster
ceasmallr
WGBS
BS-seq
Klone
Bismark

Trimming and QC - C.virginica CEASMALLR FastQs Using fastp BBTools and FastQC-MultiQC
2024
trimming
FastQC
fastp
BBTools
MultiQC
Crassostrea virginica
WGBS
BS-seq
bisulfite
Eastern oyster
ceasmallr

Taxonomic Classification - Trinity Assembly of Unmapped P.helianthoides RNA-seq Using DIAMOND BLASTx and MEGAN6 on Mox
2024
DIAMOND
BLASTx
MEGAN6
Pycnopodia helianthoides
sunflower sea star
Mox

FastQ QC - M.trossulus WGBS from Psomagen Using FastQC and MultiQC
2024
Mytilus trossulus
WGBS
Psomagen
FastQC
MultiQC
mussel
project-mytilus-methylation

Trimming and QC - M.trossulus WGBS Using fastp and FastQC-MultiQC
2024
Mytilus trossulus
WGBS
fastp
FastQC
MultiQC
bay mussel
project-mytilus-methylation
trimming
Data Wrangling - GO to GOslims for Graces Dermasterias imbricata Project from Trinity Assembly Genes
2024
Dermasterias imbricata
GO
GOslim
gene ontology
Trinity
Leather star
Data Wrangling - GO to GOslims for Graces Pycnopodia helianthoides Project from Trinity Assembly Genes
2024
GO
gene ontology
GOslim
Pycnopodia helianthoides
Trinity
Sunflower sea star

Sequencing Read Taxonomic Classification - Unmapped P.helanthoides RNA-seq Using DIAMOND-BLASTx and MEGAN6 on Mox
2024
MEGAN6
RNAseq
DIAMOND
BLASTx
Pycnopodia helianthoides
sunflower sea star
Mox
Data Received - Manila clam Supplemental sRNA-seq Data from Azenta
2024
sRNA-seq
Manila clam
Ruditapes philippinarum
30-1035633055
30-1035633055-TS01
Data Received
Data Received - Mytilus trossulus WGBS Data from Psomagen
2024
Mytilus trossulus
bay mussel
WGBS
Psomagen
AN00021156
BS-seq
bisulfite sequencing
Data Received
Data Wrangling - Get GOslims from GO IDs for Graces Pycnopodia helianthoides Project
2024
Pycnopodia helianthoides
sunflower sea star
gene ontology
GO
GOslim
data wrangling
DNA Isolation Quantification and Gel - Lake Trout Liver Samples
2024
DNA isolation
Qubit
DNA quantification
agarose gel
lake trout
Salvelinus namaycush
liver
Reverse Transcription - C.gigas Lifestage Carryover RNA
2024
reverse transcription
cDNA
GoScript
Crassostrea gigas
Pacific oyster
RNA
project-gigas-carryover
lifestage_carryover
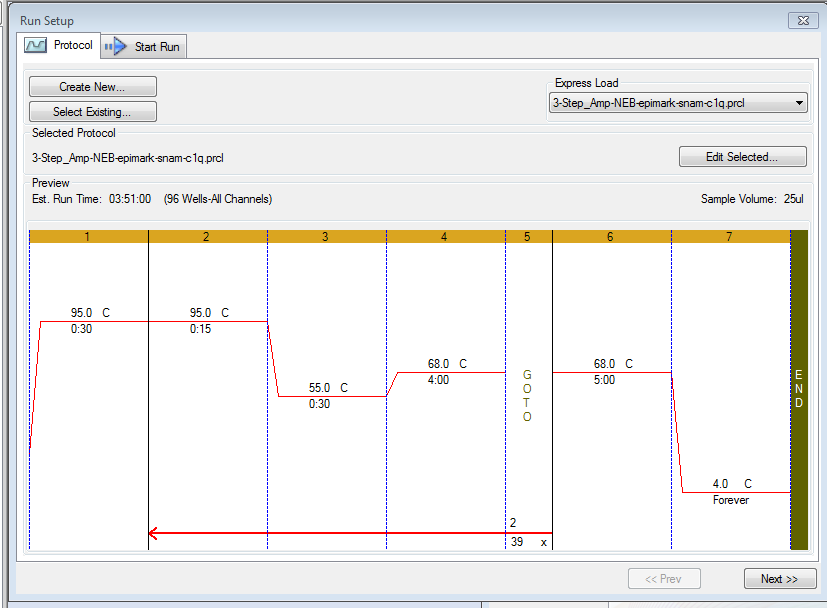
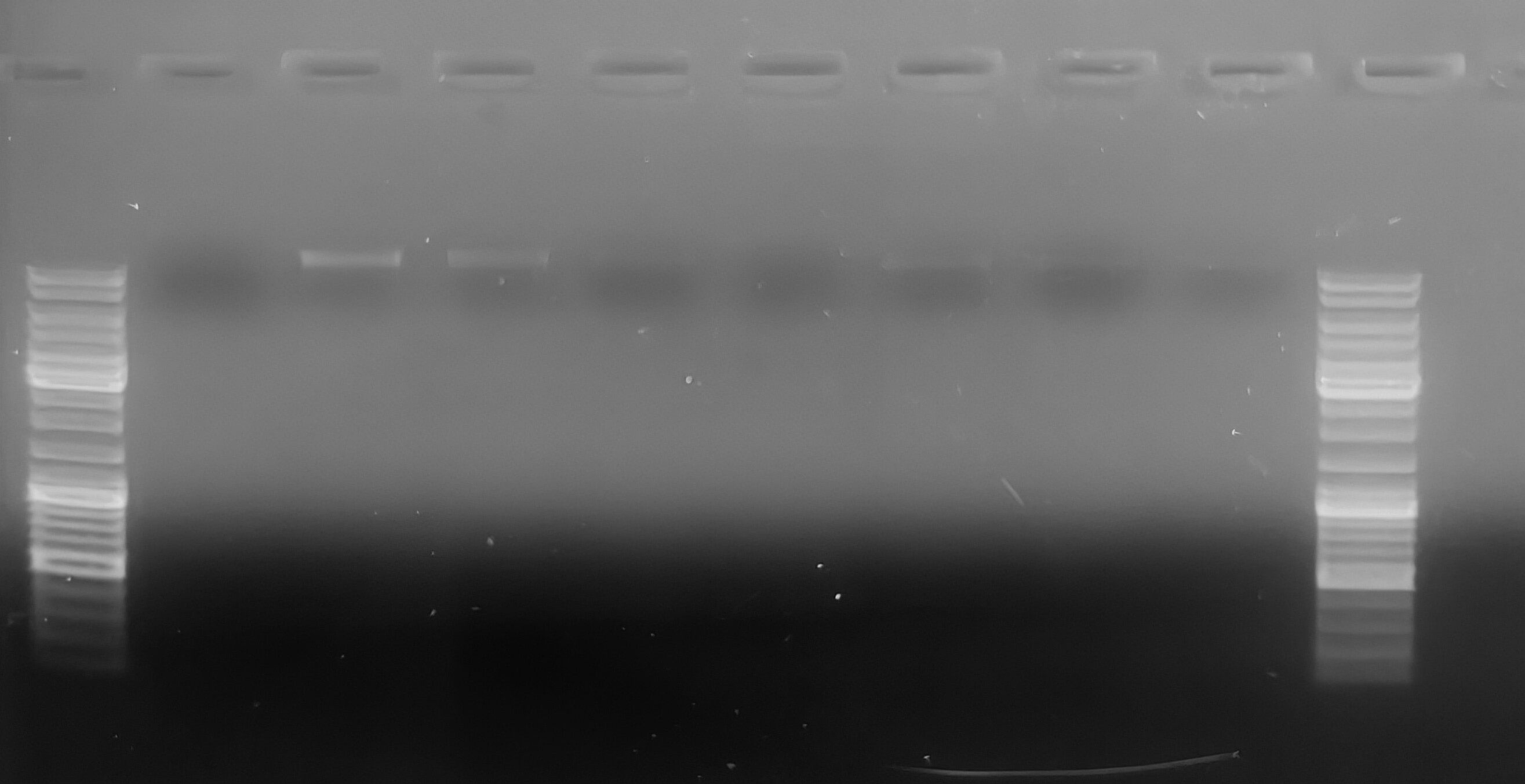
PCR - Lake Trout C1q Primers with Bisulfite-treated gDNA
2024
C1q
Lake trout
Salvelinus namaycush
PCR
bisulfite
gDNA
liver
gel
Data Wrangling - Generate GTF from A.pulchra Genome GFF Using gffread
2024
coral
Acropora pulchra
genome
GFF
GTF
gffread
E5
timeseries_molecular
Genome Indexing - A.pulchra Genome Using HISAT2
2024
coral
Acropora pulchra
HISAT2
E5
timeseries_molecular
RNA-seq Alignment - A.pulchra RNA-seq Alignments Using HISAT2 and StringTie for Azenta Project 30-1047560508
2024
30-1047560508
RNA-seq
E5
timeseries_molecular
HISAT2
StringTie
alignment
Acropora pulchra
coral
FastQC Trimming and QC - A.pulchra RNA-seq from Azenta Project 30-1047560508 Using fastp
2024
RNA-seq
30-1047560508
Azenta
coral
Acropora pulchra
fastp
FastQC
MultiQC
trimming
E5
timeseries_molecular
FastQ QC - A.pulchra RNA-seq from Azenta Project 30-1047560508
2024
RNA-seq
30-1047560508
Azenta
coral
Acropora pulchra
FastQC
MultiQC
timeseries_molecular
E5
-sRNA-seq-Data-from-Azenta-Project-30-1035633055/20241001-rphi-rSNAseq-checksums-01.png)
Data Received - R.philippinarum (Manilla clam) sRNA-seq Data from Azenta Project 30-1035633055
2024
Data Received
sRNA-seq
30-1035633055
Ruditapes philippinarum
Manila clam
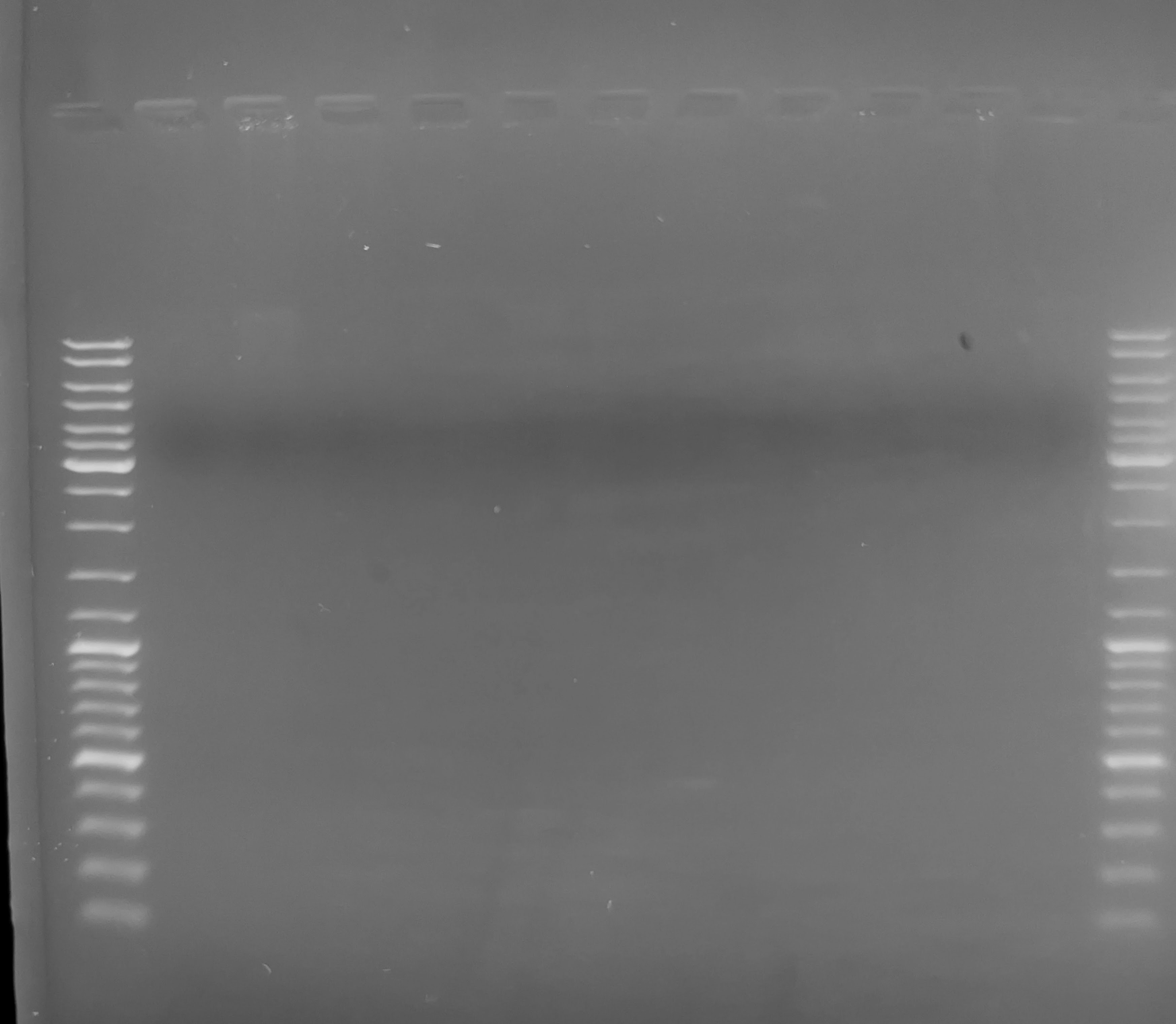
Agarose Gel - gDNA Integrity Check of Lake Trout Liver gDNA from 20240712
2024
agarose gel
gDNA
Lake trout
liver
Salvelinus namaycush
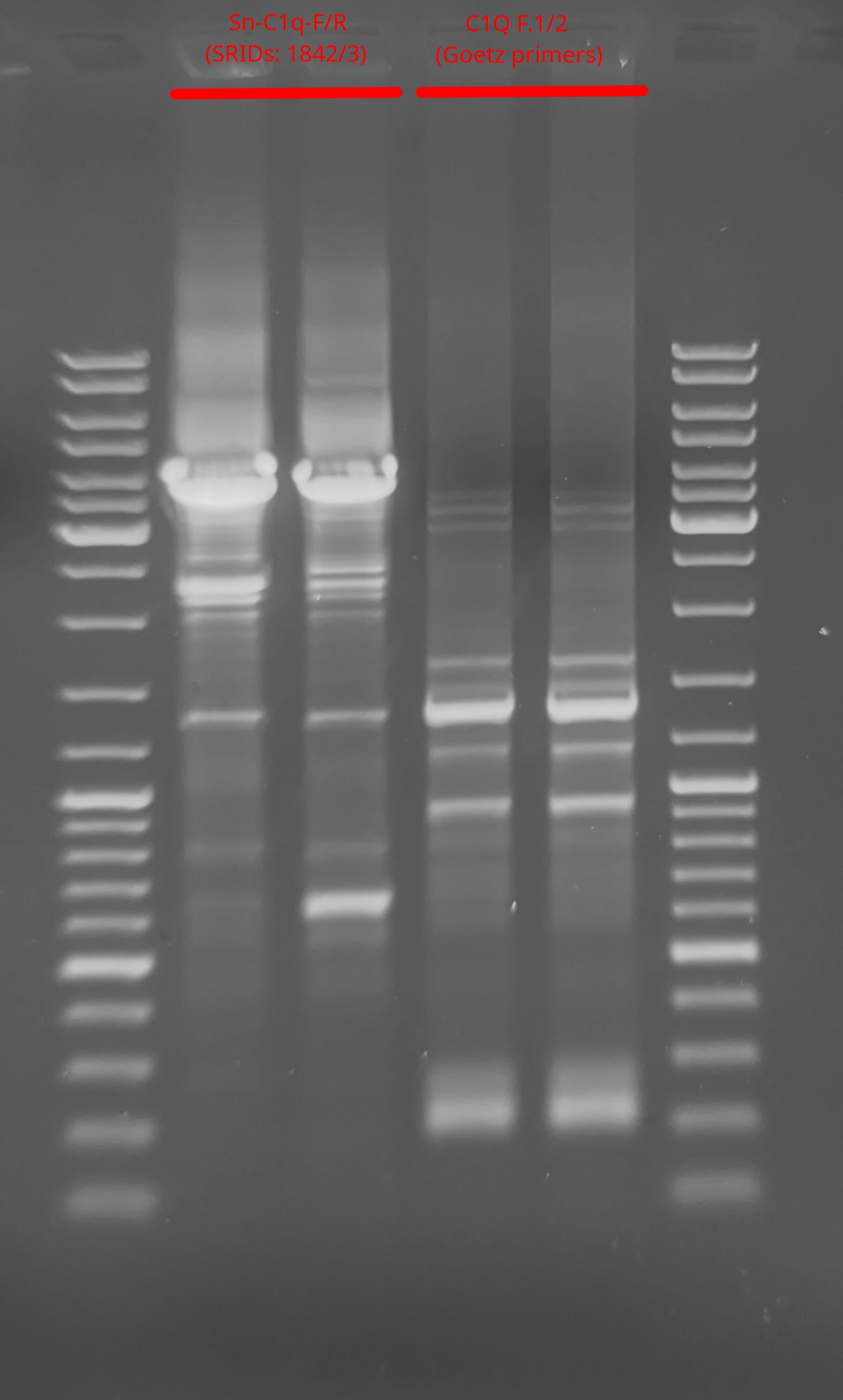
PCR - Lake Trout C1Q with Liver gDNA
2024
PCR
Lake trout
C1q
CFX Connect
EpiMark Hot Start Taq
gDNA
liver
Salvelinus namaycush
Primer Design - Lake Trout C1q Gene Sequencing Primers Using Primer3
2024
C1q
lake trout
Salvelinus namaycush
Primer3
primer design
EMBOSS
pyfaidx
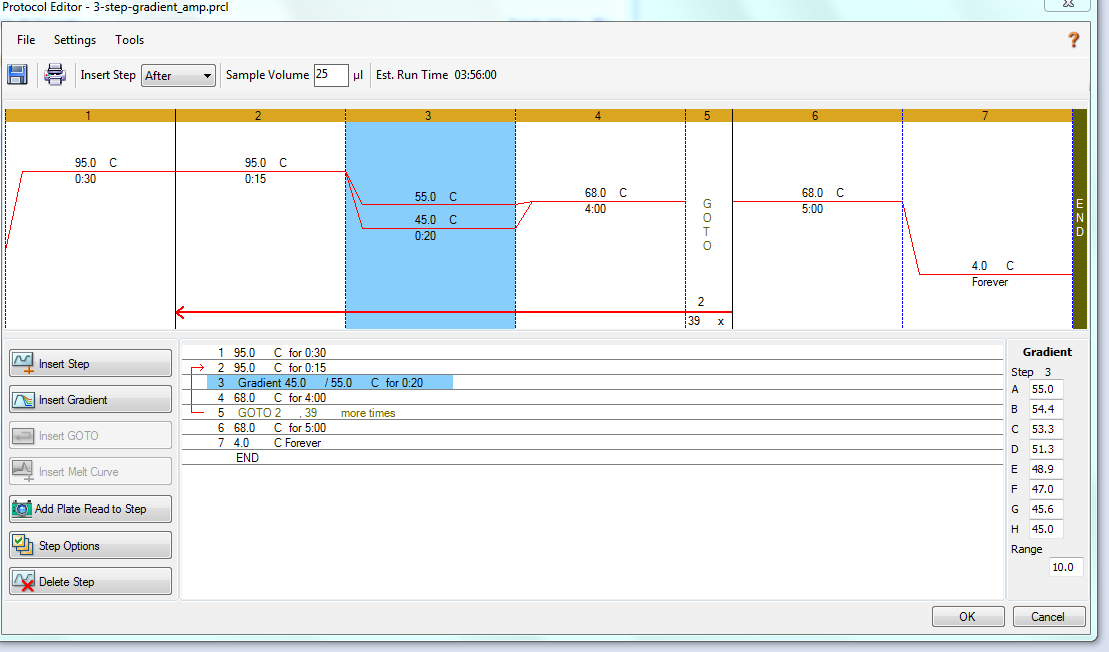
Bisulfite PCR - Second Primer Annealing Gradient Test with Lake Trout Bisulfite-treated DNA
2024
bisulfite
BS-PCR
PCR
DNA
Lake troute
Salvelinus namaycush
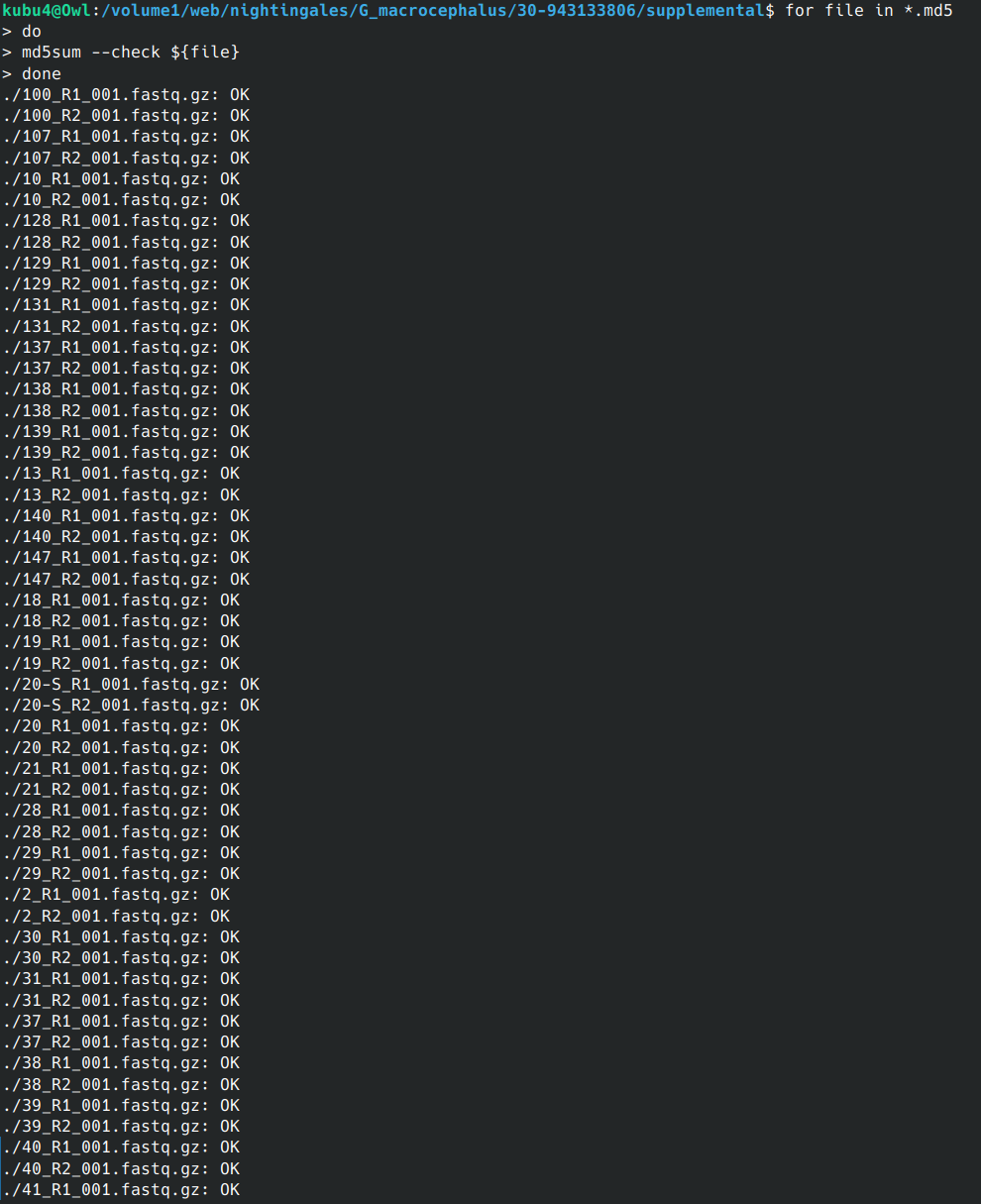
Data Received - G.macrocephalus Supplemental RNA-seq Data from Azenta Project 30-943133806
Data Received
2024
Gadus macrocephalus
Pacific cod
RNA-seq
Azenta

DNA and RNA Isolations - Gadus macrocephalus Blood
2024
DNA isolation
RNA isolation
Gadus macrocephalus
Pacific cod
blood
Zymo Quick-DNA/RNA Miniprep
Qubit 3.0
Qubit DNA BR Assay
Qubit RNA HS Assay
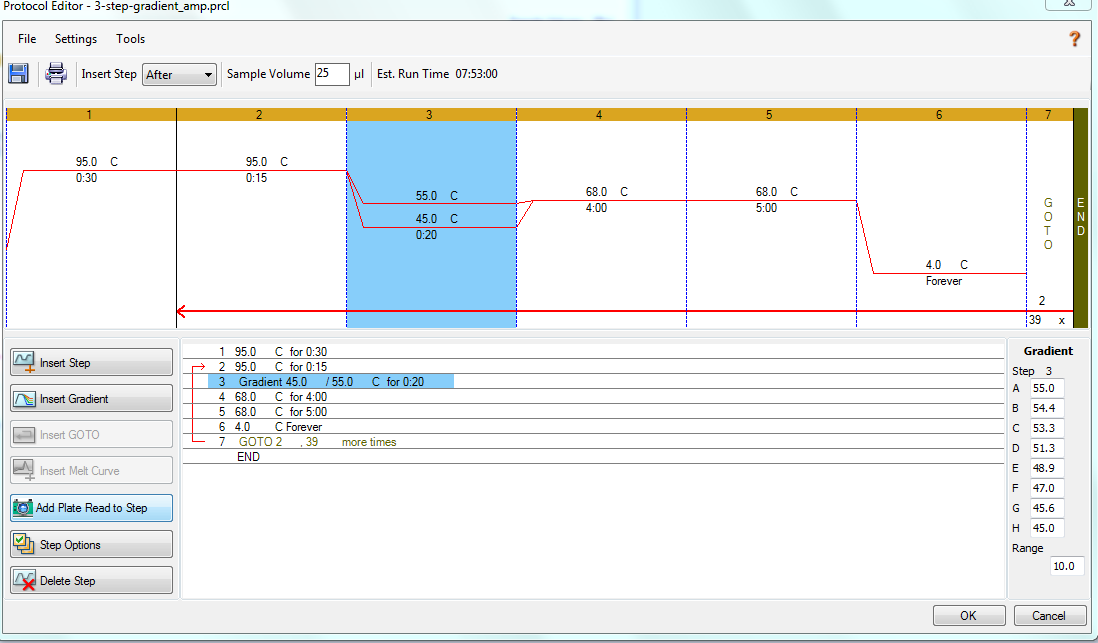
Bisulfite PCR - Testing Primer Annealing Gradient with Lake Trout Bisulfite-treated DNA
2024
BS-PCR
bisulfite
PCR
DNA
Lake trout
Salvelinus namaycush

Bisulfite Conversion - Lake Trout gDNA Using EZ DNA Methylation Lightning Kit
2024
lake trout
Salvelinus namaycush
bisulfite
EZ DNA Methylation Lightning Kit

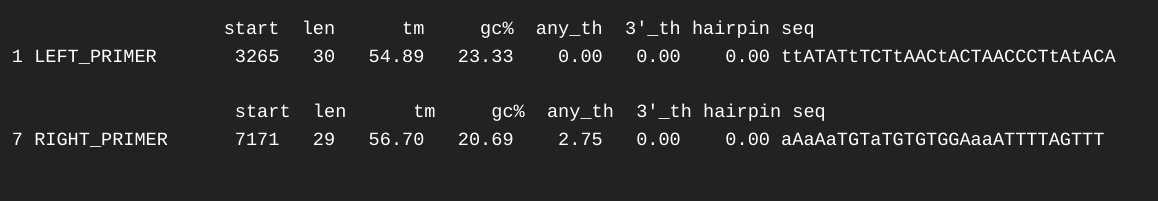
Primer Design - Lake Trout C1q Gene Bisulfite Sequencing Primers Using Primer3
2024
C1q
lake trout
Salvelinus namaycush
Primer3
primer design

Samples Submitted - Macs Manilla Clam Egg Samples for sRNA-seq at Azenta
2024
Samples Submitted
Azenta
sRNA-seq
Manila clam
Lajonkairia lajonkairii
Ruditapes philippinarum
eggs
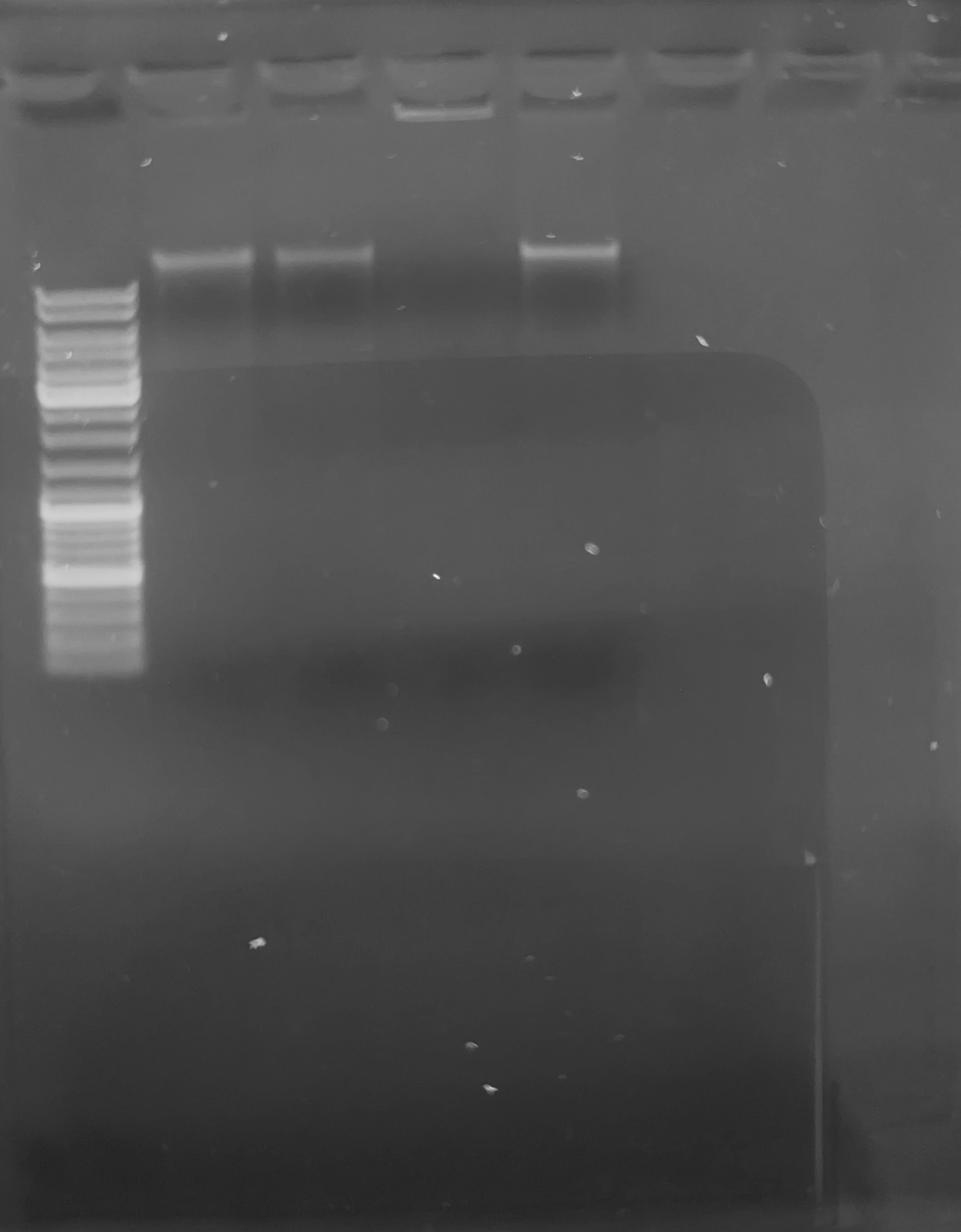
DNA Isolation Quantification and Integrity Assessment - Pacific Cod Blood Samples Test
2024
Pacific cod
Gadus macrocephalus
DNA isolation
DNA quantification
Qubit
DNA BR Assay
agarose gel
DNA Isolation and Quantification - Pacific Cod Blood
2024
Pacific cod
Gadus macrocephalus
blood
DNA isolation
DNA quantification
Qubit
DNA BR assay

DNA Isolation and Quantification - Lake Trout Liver
DNA isolation
2024
lake trout
Salvelinus namaycush
Qubit
DNA BR assay
Samples Received - L.staminea Histology Slides for Mac
2024
Leukoma staminea
littleneck clam
histology
slides
Samples Received


RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-lifestage-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA HS assay
Qubit
RNA isolation
RNA quantification
Directzol RNA miniprep
RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-lifestage-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA HS assay
Qubit
RNA isolation
Directzol RNA Miniprep
RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-lifestage-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA HS assay
Qubit
RNA isolation
Directzol RNA Miniprep

RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-lifestage-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA HS assay
Qubit
RNA isolation
Directzol RNA Miniprep

RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-lifestage-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA HS assay
Qubit
RNA isolation
Directzol RNA Miniprep

RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-gigas-carryover
Crassostrea gigas
Pacific oyster
adult
seed
spat
Directzol RNA Miniprep
Qubit
RNA HS Assay
RNA Isolation and Quantification - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-gigas-carryover
Crassostrea gigas
Pacific oyster
adult
seed
spat
Directzol RNA Miniprep
Qubit
RNA HS assay

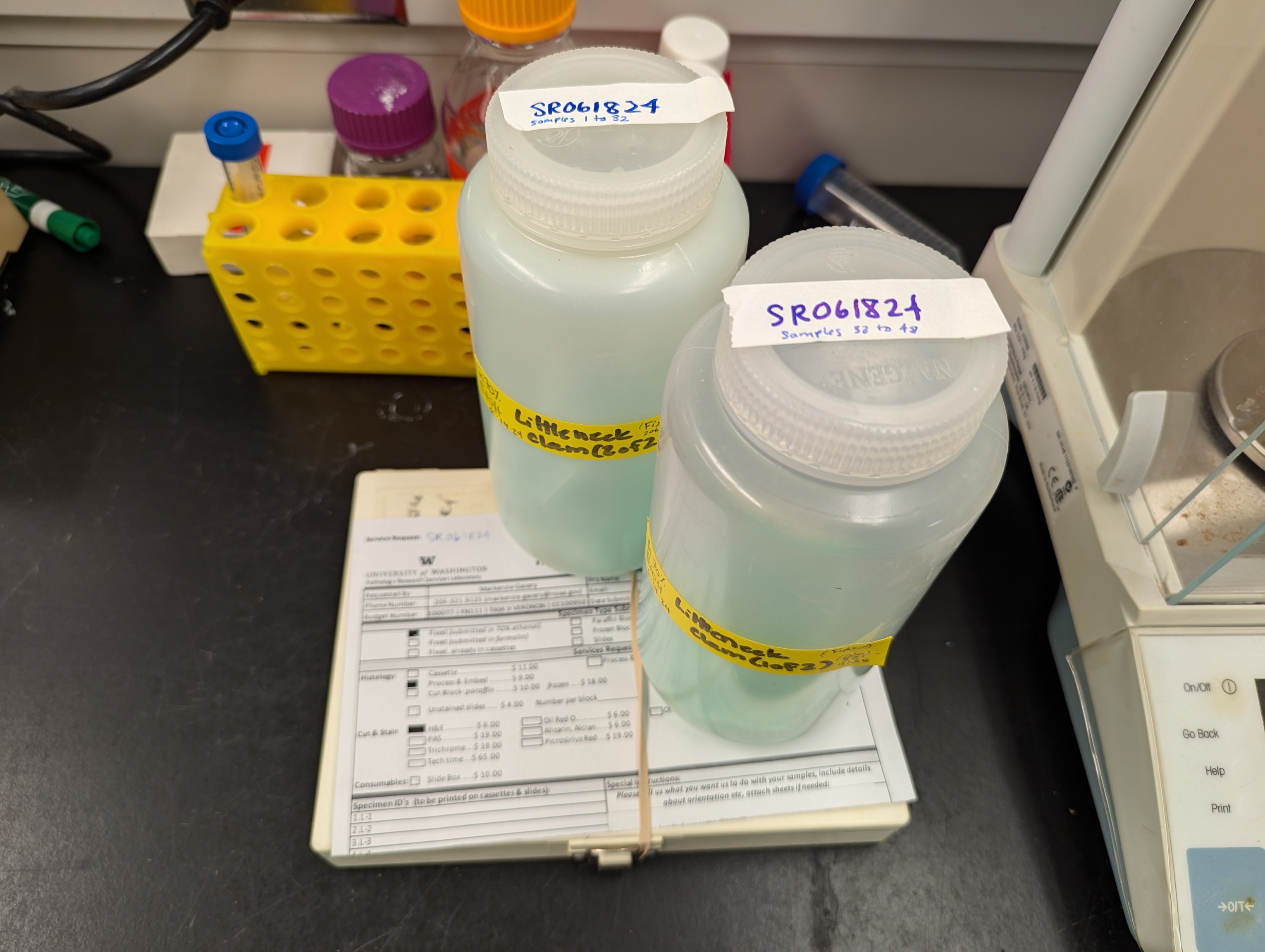
Samples Submitted - Littleneck Clam Histology Cassettes to UW Pathology Research Services Laboratory
2024
histology
littleneck clam
Pathology Research Services Laboratory
Samples Submitted
RNA Isolation - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-gigas-carryover
Directzol RNA Miniprep
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
RNA Quantification - C.gigas RNA Lifestage Carryover from 20240615 and 20240617
2024
project-gigas-carryover
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
Qubit
RNA HS assay
RNA Isolation - C.gigas Lifestage Carryover Seed Juvenile and Adult
2024
project-gigas-carryover
Directzol RNA Miniprep
Crassostrea gigas
Pacific oyster
seed
adult
juvenile
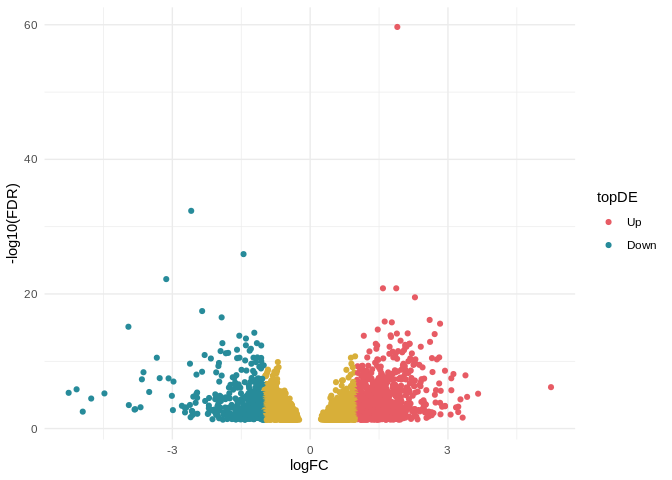
Differential Gene Expression - G.macrocephalus RNA-seq 9C vs 16C Using edgeR
edgeR
Pacific cod
RNA-seq
differential gene expression
2024
Gadus macrocephalus

Samples Received - Lajonkairia lajonkairii Egg Samples from Mackenzie Gavery
Lajonkairia lajonkairii
manilla clam
2024
Samples Received
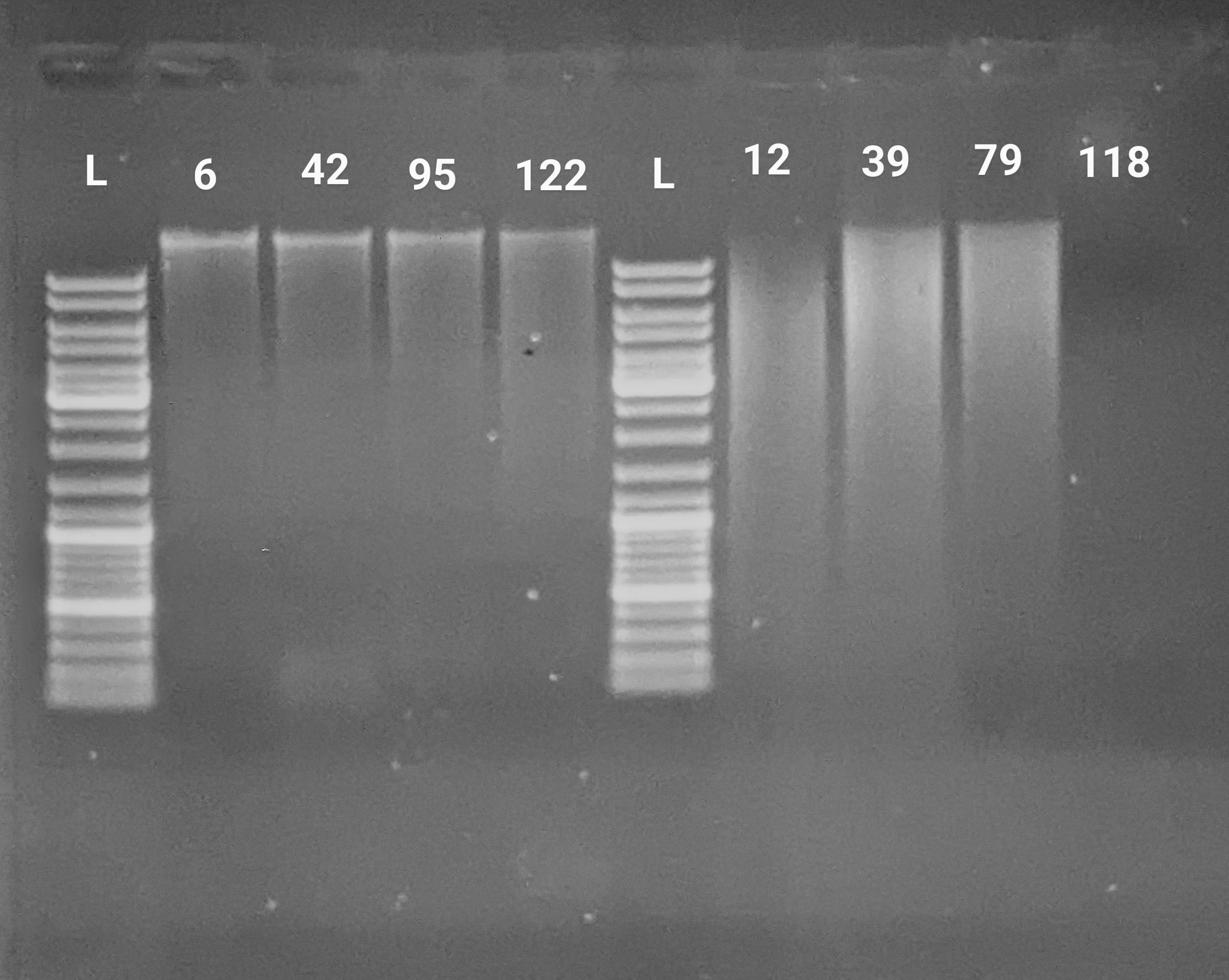
Agarose Gel - G.macrocephalus gDNA from 20240521
agarose
gDNA
Gadus macrocephalus
Pacific cod
gel
2024
DNA Isolation - Pacific cod liver
DNA isolation
Pacific cod
liver
Gadus macrocephalus
DNA quantification
Qubit
2024
Samples Submitted - Pacific Cod 40 Tissue Samples for WGBS at Psomagen
psomagen
WGBS
2024
Pacific cod
Gadus macrocephalus
BS-seq
bisulfite
Samples Submitted
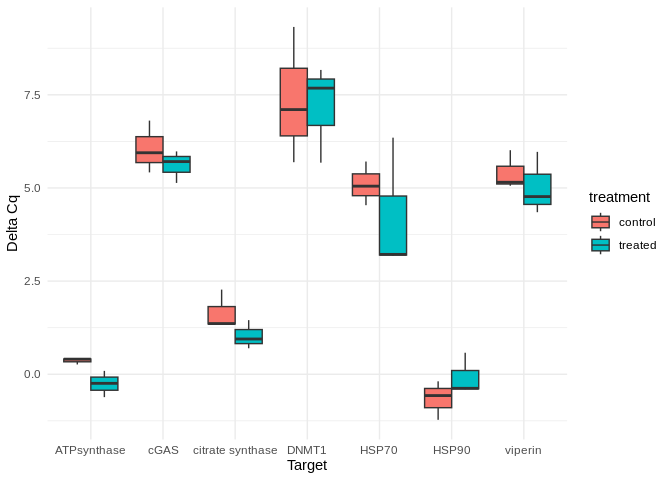
qPCR Analysis - C.gigas Lifestages Carryover from 20240325
2024
qPCR
Crassotrea gigas
Pacific oyster
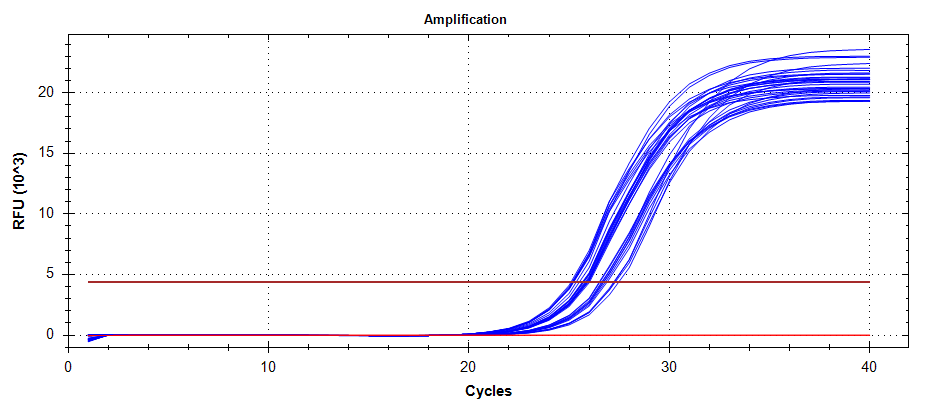
qPCRs - C.gigas Lifestage Carryover cDNA
qPCR
SsoFast
CFX Connect
HSP70
HSP90
GAPDH
VIPERIN
ATPsynthase
cGAS
DMNT1
citrate synthase
Crassostrea gigas
Pacific oyster
cDNA
RNA Isolation - C.gigas Lifestage Carryover Seed and Spat
RNA isolation
Crassostrea gigas
Lifestage carryover
Pacific oyster
2024

Reverse Transcription - C.gigas Lifestage Carryover Seed and Spat
reverse transcription
Lifestage carryover
Crassostrea gigas
Pacific oyster
cDNA
M-MLV
2024
SRA Submission - CEABIGR WGBS and RNA-seq
SRA
CEABIGR
Crassostrea virginica
Eastern oyster
RNAseq
WGBS
2024


Data Received - Pacific cod RNA-seq Azenta Project 30-943133806
Azenta
RNA-seq
2024
Pacific cod
Gadus macrocephalus
30-943133806
Data Received
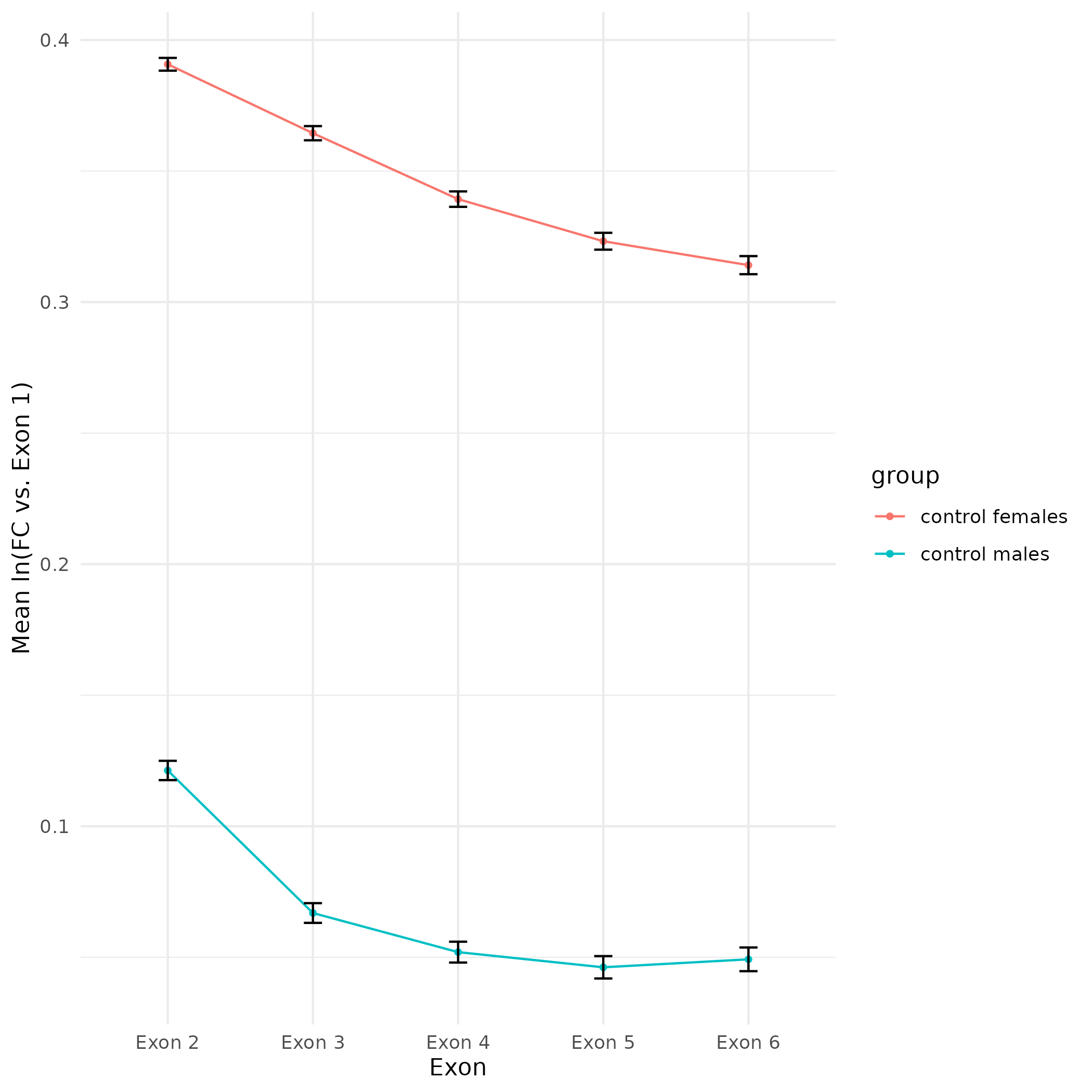
Figure Updates - CEABIGR Spurious Transcription Calcs and Plotting Using Exon Sum Threshold
CEABIGR
2024
plot
Eastern oyster
Crassostrea virginica
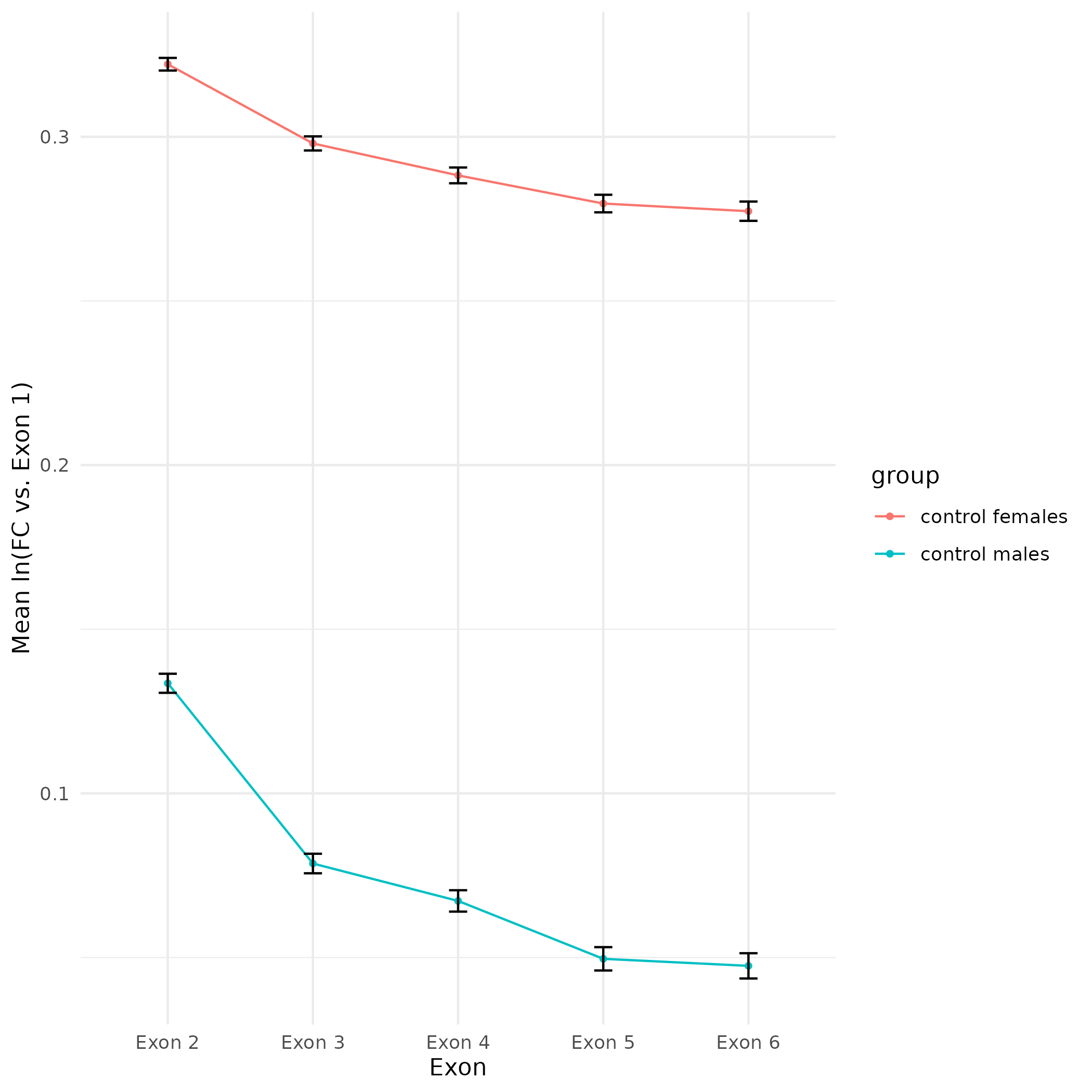
Data Exploration - CEABIGR Spurious Transcription Calculations and Plotting
2024
CEABIGR
plot
Eastern oyster
Crassostrea virginica
Samples Submitted - Gadus macrocephalus liver tissues to Azenta for RNA-seq
2023
Gadus macrocephalus
Pacific cod
Azenta
RNA-seq
Samples Submitted
liver
spleen
gill
blood
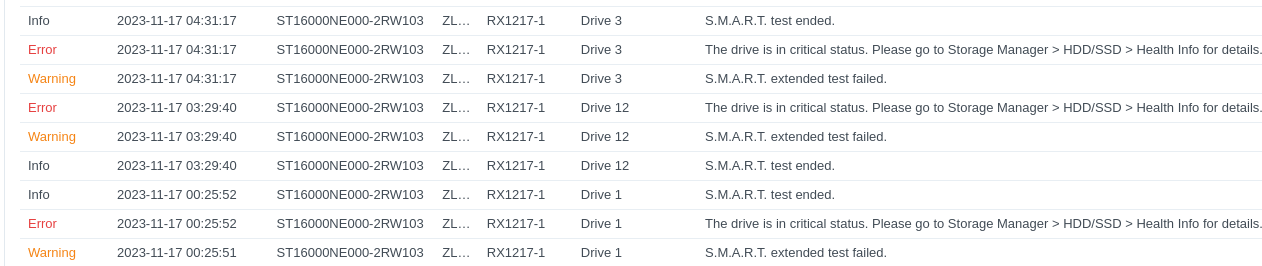
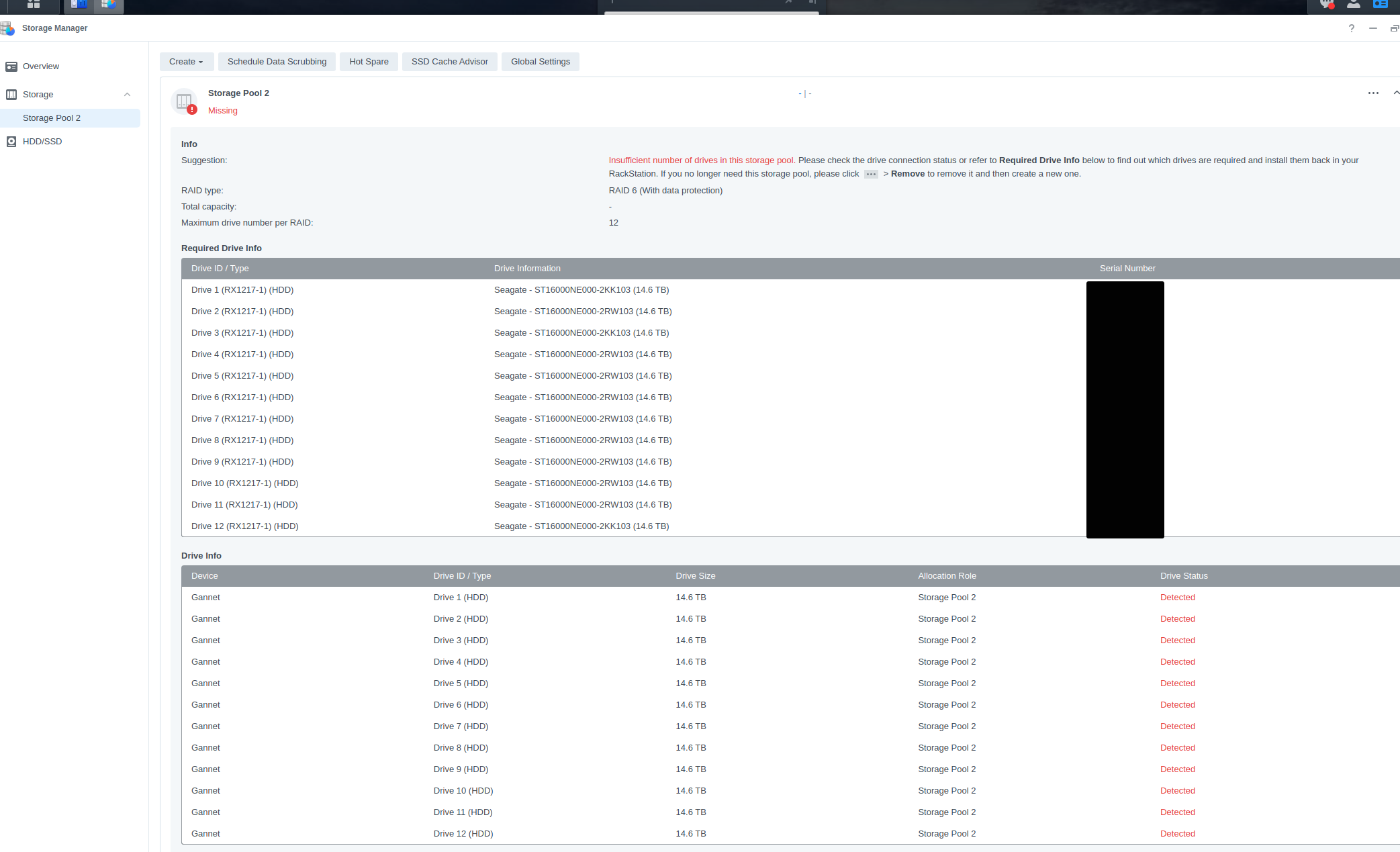
Computer Maintenance - Replace Failing HDDs in Synology RX1217 Expansion Unit on Gannet
gannet
synology
RX1217
HDDs
2023
computer maintenance
Transcriptome Annotation - M.magister De Novo Transcriptome Assembly for DuMOAR Project Using DIAMOND BLASTx on Mox
mox
DIAMOND
BLASTx
transcriptome
2023
DuMOAR
Dungeness crab
Metacarcinus magister
Transcriptome Annotation - M.magister De Novo Transcriptome Assembly Using Trinotate on Mox
mox
2023
Metacarcinus magister
Dungenss crab
Trinotate
transcriptome
annotation
qPCR Analysis - C.gigas Matt George PolyIC Diploids
qPCR
CFX Connect
Crassostrea gigas
diploid
Pacific oyster
2023
Miscellaneous
Transcript Identification and Alignments - C.virginica RNAseq with NCBI Genome GCF_002022765.2 Using Hisat2 and Stringtie on Mox Again
hisat2
mox
Crassostrea virginica
stringtie
RNAseq
Eastern oyster
2023
Miscellaneous
RNA Quantification - C.gigas PolyIC Diploid MgCl2
polyIC
Crassostrea gigas
Pacific oyster
2023
Miscellaneous
Read Extractions - M.magister MEGAN FastA to FastQ Arthropoda and Unassigned Reads
Metacarcinus magister
Dungeness crab
MEGAN
2023
DuMOAR
Read Extractions - M.magister MEGAN Arthropoda and Unassigned Reads to FastA
Metacarcinus magister
Dungeness crab
MEGAN
2023
DuMOAR
qPCR - C.gigas cDNA Primer Tests for Noah’s Heat-Mechanical Stress Project
qPCR
Crassostrea gigas
cDNA
Noah
Pacific oyster
2023
Reverse Transcription - C.gigas RNA from Noah’s Heat-Mechanical Stress Project
reverse transcription
Crassostrea gigas
RNA
Pacific oyster
2023
File Conversion - M.magister MEGANized DAA to RMA6
megan
DAA
RMA6
Metacarcinus magister
dungeness crab
2023
Miscellaneous
Trimming and QC - E5 Coral sRNA-seq Data fro A.pulchra P.evermanni and P.meandrina Using FastQC flexbar and MultiQC on Mox
flexbar
E5
trimming
FastQC
MultiQC
mox
coral
2023
Data Wrangling - P.meandrina Genome GFF to GTF Using gffread
gffread
jupyter
Pocillopora meandrina
cora
E5
GFF
GTF
2023
FastQ QC and Trimming - E5 Coral RNA-seq Data for A.pulchra P.evermanni and P.meandrina Using FastQC fastp and MultiQC on Mox
mox
FastQC
fastp
MultiQC
E5
coral
Acropora pulchra
Porites evermanni
Pocillopora meandrina
2023
Data Received - Coral RNA-seq Data from Azenta Project 30-789513166
30-789513166
RNA-seq
Azenta
coral
Putnam
2023
Data Received
lncRNA Identification - P.generosa lncRNAs using CPC2 and bedtools
lncRNA
Panopea generosa
Pacific geoduck
CPC2
bedtools
2023
Transcript Alignments - P.generosa RNA-seq Alignments for lncRNA Identification Using Hisat2 StingTie and gffcompare on Mox
gffcompare
mox
Panopea generosa
Pacific geoduck
lncRNA
StringTie
Hisat2
2023
Miscellaneous
Genome Indexing - M.capitata NCBI GCA_006542545.1 with HiSat2 on Mox
hisat2
Montipora capitata
coral
GCA_006542545.1
mox
2023
Miscellaneous
SRA Data - Coral SRA BioProject PRJNA744403 Download and QC
PRJNA744403
SRA
NCBI
fastp
FastQC
MultiQC
coral
metagenomics
2023
Miscellaneous
FastQ Trimming and QC - C.virginica Larval BS-seq Data from Lotterhos Lab and Part of CEABIGR Project Using fastp on Mox
BSseq
Lotterhos
CEABIGR
Crassostrea virginica
Eastern oyster
fastp
mox
2022
Miscellaneous
Project Summary - C.virginica CEABiGR - Female vs. Male Gonad Exposed to OA
Crassostrea virginica
CEABIGR
OA
gonad
Eastern oyster
2022
Project Summary
Server Maintenance - Fix Server Certificate Authentication Issues
github
InCommon
certificate
servers
gannet
owl
eagle
2022
Computer Servicing
Differential Gene Expression - P.generosa DGE Between Tissues Using Nextlow NF-Core RNAseq Pipeline on Mox
nextflow
Panopea generosa
Pacific geoduck
RNAseq
nf-core
mox
2022
Miscellaneous
Transcript Identification and Alignments - C.virginica RNAseq with NCBI Genome GCF_002022765.2 Using Hisat2 and Stringtie on Mox
hisat2
stringtie
Crassostrea virginica
Eastern oyster
RNAseq
GCF_002022765.2
2022
Miscellaneous
Trimming - Additional 20bp from C.virginica Gonad RNAseq with fastp on Mox
fastp
mox
Crassostrea virginica
RNAseq
trimming
Eastern oyster
2022
Miscellaneous
RNAseq Alignment - C.virginica Adult OA Gonad Data to GCF_002022765.2 Genome Using HISAT2 on Mox
HISAT2
Crassostrea virginica
RNAseq
Mox
Eastern oyster
2022
Miscellaneous
Data Wrangling - C.virginica Gonad RNAseq Transcript Counts Per Gene Per Sample Using Ballgown
data wrangling
Crassostrea virginica
eastern oyster
R
ballgown
2022
Miscellaneous
Data Received - C.virginica OA Larvae DNA Methylation FastQs from Lotterhos Lab
Crassostrea virginica
Eastern oyster
larvae
DNA methylation
data received
2022
Data Received
RNA Isolation - O.nerka Berdahl Tissues
RNA isolation
qubit
RNA quantification
RNA
2021
Miscellaneous
Differential Transcript Expression - C.virginica Gonad RNAseq Using Ballgown
Crassostrea virginica
Eastern oyster
gonad
RNAseq
ballgown
DEG
DET
expression
2021
Miscellaneous
SNP Identification - C.bairdi Day 2 DEG Pooled Samples Using bcftools on Mox
bcftools
Tanner crab
Chionoecetes bairdi
Mox
SNP
2021
Tanner Crab RNAseq
RNAseq Alignments - C.bairdi Day 2 Infected-Uninfected Temperature Increase-Decrease RNAseq to cbai_transcriptome_v3.1.fasta with Hisat2 on Mox
hisat2
alignment
RNAseq
Tanner crab
Chionoecetes bairdi
mox
2021
Tanner Crab RNAseq
Computer Servicing - APC SUA2200RM2U UPS Battery Replacement
battery
ups
apc
2021
Computer Servicing
Assembly Comparisons - Ostrea lurida Non-scaffold Genome Assembly Comparisons Using Quast on Swoose
swoose
quast
Ostrea lurida
Olympia oyster
2021
Olympia Oyster Genome Assembly
Trimming - O.lurida BGI FastQs with FastP on Mox
fastp
mox
Ostrea lurida
Olympia oyster
trimming
2021
Olympia Oyster Genome Sequencing
Genome Submission - Validation of Olurida_v081.fa and Annotated GFFs Prior to Submission to NCBI
2021
Olympia Oyster Genome Assembly
RepeatMasker - C.gigas Rosling NCBI Genome GCA_902806645.1 on Mox
GCA_902806645.1
mox
repeatmasker
Crassostrea gigas
Pacific oyster
2021
Miscellaneous
Singularity - RStudio Server Container on Mox
computing
Rstudio Server
mox
singularity
2021
Miscellaneous
Transcriptome Assembly - C.bairdi Transcriptome v4.0 Using Trinity on Mox
mox
Tanner crab
Chionoecetes bairdi
Trinity
transcriptome
assembly
RNAseq
2021
Tanner Crab RNAseq
Transcriptome Assembly - Hematodinium Transcriptomes v1.6 and v1.7 with Trinity on Mox
Tanner crab
hematodinium
mox
trinity
Chionoecetes bairdi
2021
Miscellaneous
Samples Submitted - M.magister MBD-BSseq Libraries to Univ. of Oregon GC3F
Metacarcinus magister
sequencing
Dungeness crab
Cancer magister
2021
Miscellaneous
ENA Submission - Ostrea lurida draft genome Olurida_v081.fa
2020
Olympia Oyster Genome Sequencing
Data Received - C.bairdi RNAseq Data from Genewiz
2020
Data Received
Tanner Crab RNAseq
Data Wrangling - Renaming, Splitting, and Feature Counts of Updated Pgenerosa_v074 GenSAS Merged GFF
2019
Geoduck Genome Sequencing
Data Received - C.bairdi RNAseq Day9-12-26 Infected-Uninfected
2019
Tanner Crab RNAseq
Data Received
Genome Annotation - O.lurida 20190709-v081 Transcript Isoform ID with Stringtie on Mox
2019
Olympia Oyster Genome Sequencing
Sample Submission - Tanner Crab Infected vs Uninfected RNAseq
2019
Tanner Crab RNAseq
Samples Submitted
Data Management - Create C.virginica Bisulfite Genome with Bismark on Mox
2019
Crassostrea virginica
eastern oyster
mox
bismark
Metagenome Assembly - P.generosa Water Sample Trimmed HiSeqX Data Using Megahit on Mox
Panopea generosa
geoduck
megahit
mox
metagenome
assembly
2019
Miscellaneous
Repeat Library Construction - P.generosa RepeatModeler v1.0.11
emu
repeatmodeler
jupyter notebook
Panopea generosa
geoduck
Pgenerosa_v070.fa
2018
Geoduck Genome Sequencing
qPCR - Relative mitochondrial abundance in C.gigas diploids and triploids subjected to acute heat stress via COX1
qPCR
Crassostrea gigas
Pacific oyster
CFX Connect
SsoFast EVAGreen
COX1
2018
Miscellaneous
qPCR - Geoduck gonad cDNA with vitellogenin primers
qPCR
CFX
2x SsoFast EVAgreen
geoduck
vtg
vitellogenin
Panopea generosa
gonad
2018
Annotation - Olurida_v081 MAKER on Mox
Ostrea lurida
Olympia oyster
mox
MAKER
genome annotation
Olurida_v081
2018
Olympia Oyster Genome Sequencing
Annotation - Geoduck Transcritpome with TransDecoder
TransDecoder
geoduck
Panopea generosa
annotation
transcriptome
mox
2018
RNA Quantification - Ronit’s C.gigas DNased RNA from 20181115
RNA quantification
Qubit 3.0
Qubit hsRNA
Crassostrea gigas
pacific oyster
diploid
triploid
dessication
DNased RNA
2018
Reverse Transcription - Ronit’s C.gigas DNased RNA from 20181115
reverse transcription
cDNA
MMLV
pacific oyster
Crassostrea gigas
diploid
triploid
dessication
2018
Repeat Library Construction - O.lurida RepeatModeler v1.0.11
emu
repeatmodeler
jupyter notebook
Ostrea lurida
olympia oyster
2018
Olympia Oyster Genome Sequencing
qPCR – C.gigas primer and gDNA tests with 18s and EF1 primers
2018
BB15
BB16
cfx connect
Cg_18s_1644_F
Cg_18s_1750_R
Crassostrea gigas
EF1_qPCR_3'
EF1_qPCR_5'
Pacific oyster
qPCR
SRID 1168
SRID 1169
SRID 309
SRID 310
SsoFast EvaGreen Supermix
RNA Isolation - Tanner Crab Hemolymph Pellet in RNAlater using TriReagent
2018
Chionoecetes bairdi
Qubit 3.0
Qubit RNA HS
RNA
RNA isolation
RNA quantification
tanner crab
TriReagent
SRA Submission - Olymia oyster Whole Genome BS-seq Data
2018
Olympia oyster reciprocal transplant
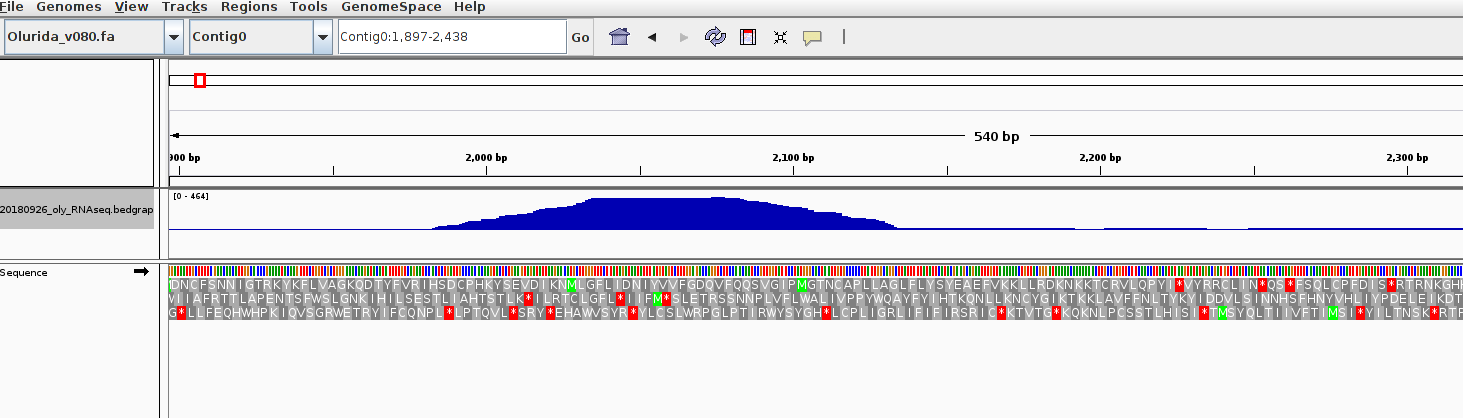

TrimGalore/FastQC/MultiQC - C.virginica Oil Spill MBDseq Concatenated Sequences
2018
LSU C.virginica Oil Spill MBD BS Sequencing
DNA Methylation Analysis - Olympia oyster BSseq MethylKit Analysis
2018
bismark
BSseq
methylation
methylkit
Olympia oyster
Ostrea lurida
FastQC/MultiQC/TrimGalore/MultiQC/FastQC/MultiQC - O.lurida WGBSseq for Methylation Analysis
2018
BSseq
FASTQC
mox
multiqc
olympia oyster
Ostrea lurida
TrimGalore!
trimming
Genome Annotation – Olympia oyster genome annotation results #02
2018
Olympia Oyster Genome Sequencing
Genome Annotation - Olympia oyster genome annotation results #01
2018
Olympia Oyster Genome Sequencing
Genome Annotation - Olympia oyster genome using WQ-MAKER Instance on Jetstream
2018
Olympia Oyster Genome Sequencing
annotation
Atmosphere
Cyverse
genome
JetStream
maker
olympia oyster
Ostrea lurida
wq-maker
Xsede
Mox – Over quota: Olympia oyster genome annotation progress (using Maker 2.31.10)
2018
Olympia Oyster Genome Sequencing
Library Construction - Geoduck Water Filter Metagenome with Nextera DNA Flex Kit (Illumina)
2018
bioanalyzer
geoduck
library prep
metagenomics
nextera
Panopea generosa
Qubit 1x dsDNA HS Assay Kit
Qubit 3.0
Software Installation - RepeatMasker v4.0.7 on Emu/Roadrunner
2018
emu
RepeatMasker
roadrunner
TE
transposable elements
BS-seq Mapping - Olympia oyster bisulfite sequencing: TrimGalore > FastQC > Bismark
2018
BS-seq Libraries for Sequencing at Genewiz
MBD Enrichment for Sequencing at ZymoResearch
Data Management - Geoduck Phase Genomics Hi-C Data
2018
Geoduck Genome Sequencing
Samples Received
Kmer Estimation – Kmergenie Tweaks on Geoduck Sequence Data
2018
Geoduck Genome Sequencing
geoduck
kmer
kmer genie
mox
Panopea generosa
Kmer Estimation - Kmergenie on Geoduck Sequence Data (default settings)
2018
Geoduck Genome Sequencing
geoduck
kmer
kmer genie
mox
Panopea generosa
Data Management - SRA Submission LSU C.virginica Oil Spill MBD BS-seq Data
2018
LSU C.virginica Oil Spill MBD BS Sequencing
TrimGalore/FastQC/MultiQC - Trim 10bp 5’/3’ ends C.virginica MBD BS-seq FASTQ data
2018
Crassostrea virginica
Eastern oyster
FASTQC
MBD-Seq
multiqc
TrimGalore!
ZymoResearch
TrimGalore/FastQC/MultiQC - 2bp 3’ end Read 1s Trim C.virginica MBD BS-seq FASTQ data
2018
Crassostrea virginica
Eastern oyster
FASTQC
MBD-Seq
multiqc
TrimGalore!
ZymoResearch
Genome Assembly - SparseAssembler Geoduck Genomic Data, kmer=101
2018
Geoduck Genome Sequencing
Assembly - Geoduck NovaSeq using SparseAssembler (failed)
2018
Geoduck Genome Sequencing
assembly
geoduck
NovaSeq
Panopea generosa
SparseAssembler
Progress Report - Titrator
2018
CRM
Rondolino
T5
T5 Excellence
titrator
NovaSeq Assembly - The Struggle is Real - Real Annoying!
2018
Geoduck Genome Sequencing
AllPaths-LG
assembly
geoduck
jr-assembler
meraculous
mox
NovaSeq
Panopea generosa
SOAPdenovo2
NovaSeq Assembly - Trimmed Geoduck NovaSeq with Meraculous
2018
Geoduck Genome Sequencing
assembly
geoduck
Illumina
kmer
kmer genie
meraculous
NovaSeq
Panopea generosa
roadrunner
Adapter Trimming and FASTQC - Illumina Geoduck Novaseq Data
2018
Geoduck Genome Sequencing
FASTQC
geoduck
Illumina
multiqc
NovaSeq
Panopea generosa
trim galore
trimming
Software Install - 10x Genomics Supernova on Mox (Hyak)
2018
10x genomics
bcl2fastq2
hyak
mox
supernova
Assembly Comparisons – Oly Assemblies Using Quast
2018
Olympia Oyster Genome Sequencing
assembly
jupyter notebook
Olympia oyster
Ostrea lurida
QUAST
DNA Quantification - MspI-digested Crassostrea virginica gDNA
2018
Crassostrea virginica
Eastern oyster
Qubit 3.0
Qubit dsDNA BR
Phenol:Chloroform Extractions and EtOH Precipitations - MspI Digestions of C.virginica DNA from Earlier Today
2018
Crassostrea virginica
Eastern oyster
EtOH precipitation
phenol-chloroform cleanup
DNA Quantification - C.virginica MBD-enriched DNA
2018
Crassostrea virginica
DNA Quantification
Eastern oyster
Qubit 3.0
Qubit dsDNA BR
MBD Enrichment – Crassostrea virginica Sheared DNA Day 3
2018
Crassostrea virginica
Eastern oyster
EtOH precipitation
MBD
MBD enrichment
MethylMiner Methylated DNA Enrichment Kit
MBD Enrichment – Crassostrea virginica Sheared DNA Day 2
2018
Crassostrea virginica
Eastern oyster
MBD
MBD enrichment
MethylMiner Methylated DNA Enrichment Kit
Samples Submitted - Pulverized Geoduck Tissues to Illumina for More 10x Genomics Sequencing
2017
Geoduck Genome Sequencing
Samples Submitted
2017
Geoduck Genome Sequencing
Samples Submitted
Genome Assembly – Olympia Oyster Illumina & PacBio Using PB Jelly w/BGI Scaffold Assembly
2017
Olympia Oyster Genome Sequencing
assembly
Illumina
jupyter notebook
olympia oyster
Ostrea lurida
PacBio
PB Jelly
pbjelly
QUAST
Samples Submitted - Geoduck Tissues to Illumina for More 10x Genomics Sequencing
2017
Geoduck Genome Sequencing
Samples Submitted
geoduck
Illumina
Panopea generosa
Assembly Comparison - Oly Assemblies Using Quast
2017
Olympia Oyster Genome Sequencing
assembly
olympia oyster
Ostrea lurida
QUAST
Genome Assembly - Olympia Oyster Illumina & PacBio Using PB Jelly w/BGI Scaffold Assembly
2017
Olympia Oyster Genome Sequencing
assembly
BGI
Illumina
jupyter notebook
olympia oyster
Ostrea lurida
PacBio
PB Jelly
Genome Assembly - Olympia Oyster Illumina & PacBio Using PB Jelly w/Platanus Assembly
2017
Olympia Oyster Genome Sequencing
assembly
Illumina
jupyter notebook
olympia oyster
Ostrea lurida
PacBio
PB Jelly
Genome Assembly - Olympia oyster Illumina & PacBio reads using MaSuRCA
2017
Olympia Oyster Genome Sequencing
Genome Assembly - Olympia oyster PacBio Canu v1.6
2017
Olympia Oyster Genome Sequencing
canu
jupyter notebook
olympia oyster
Ostrea lurida
PacBio
Data Management - Convert Oly PacBio H5 to FASTQ
2017
Olympia Oyster Genome Sequencing
FASTQ
olympia oyster
Ostrea lurida
PacBio
Genome Assembly - Olympia Oyster Redundans with Illumina + PacBio
2017
Olympia Oyster Genome Sequencing
Genome Assembly - minimap/miniasm/racon Overview
2017
Olympia Oyster Genome Sequencing
Assembly Comparisons - Olympia oyster genome assemblies
2017
Olympia Oyster Genome Sequencing
docker
jupyter notebook
olympia oyster
Ostrea lurida
QUAST
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
2017
minimap
racon
miniasm
olympia oyster
Ostrea lurida
PacBio
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
2017
Olympia Oyster Genome Sequencing
jupyter notebook
miniasm
olympia oyster
Ostrea lurida
PacBio
racon
Genome Assembly - Olympia oyster PacBio minimap/miniasm/racon
2017
Olympia Oyster Genome Sequencing
jupyter notebook
minimap
olympia oyster
Ostrea lurida
PacBio
Samples Submitted - Geoduck Ctenidia to Illumina for 10x Genomics Sequencing
2017
Geoduck Genome Sequencing
Samples Submitted
10x genomics
ctenidia
geoduck
Illumina
Panopea generosa
Project Progress - Olympia Oyster Genome Assemblies by Sean Bennett
2017
Olympia Oyster Genome Sequencing
BGI
Illumina
olympia oyster
Ostrea lurida
PacBio
Sample Submission - Olympia oyster gonad RNA to Katherine Silliman @ Univ. of Chicago
2017
Samples Submitted
NF-10 22
NF-10-23
NF-10-24
NF-10-26
NF-10-28
NF-10-30
olympia oyster
Ostrea lurida
RNA
SN-10-16
SN-10-17
SN-10-20
SN-10-25
SN-10-26
SN-10-31
Data Management - Olympia oyster UW PacBio Data from 20170323
2017
Olympia Oyster Genome Sequencing
jupyter notebook
olympia oyster
Ostrea lurida
PacBio
Manuscript Writing - Submitted!
2017
Genotype-by-sequencing at BGI
Manuscript - Oly GBS 14 Day Plan
2017
Genotype-by-sequencing at BGI
Data Management – SRA Submission Oly GBS Batch Submission
2017
Genotype-by-sequencing at BGI
Computing – Oly BGI GBS Reproducibility; fail?
2017
Genotype-by-sequencing at BGI
BGI
GBS
genotype-by-sequencing
jupyter notebook
olympia oyster
Ostrea lurida
Computing - Oly BGI GBS Reproducibility Fail
2017
Genotype-by-sequencing at BGI
BGI
GBS
genotype-by-sequencing
iTools
olympia oyster
Ostrea lurida
FASTQC - Oly BGI GBS Raw Illumina Data Demultiplexed
2017
Genotype-by-sequencing at BGI
BGI
docker
FASTQC
GBS
genotype-by-sequencing
jupyter notebook
olympia oyster
Ostrea lurida
ostrich
FASTQC - Oly BGI GBS Raw Illumina Data
2017
Genotype-by-sequencing at BGI
BGI
FASTQC
GBS
genotype-by-sequencing
jupyter notebook
olympia oyster
Ostrea lurida
Data Received - Jay’s Coral RADseq and Hollie’s Geoduck Epi-RADseq
2017
Anthopleura elegantissima
docker
geoduck
jupyter notebook
Sample Submission – Geoduck gDNA for Illumina Pilot Sequencing Project
2017
Geoduck Genome Sequencing
Samples Submitted
DNA Isolation - Geoduck gDNA for Illumina-initiated Sequencing Project
2017
Geoduck Genome Sequencing
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
gDNA
gel
geoduck
Illumina
Panopea generosa
Qubit 3.0
Qubit dsDNA BR
Data Management - Replacement of Corrupt BGI Oly Genome FASTQ Files
2017
Olympia Oyster Genome Sequencing
BGI
jupyter notebook
md5
olympia oyster
Ostrea lurida
Data Management - Geoduck RRBS Data Integrity Verification
2016
geoduck
jupyter notebook
md5
Panopea generosa
RRBS
Data Received - Geoduck RRBS Sequencing Data
2016
Genewiz
geoduck
Illumina
Panopea generosa
RRBS
Sample Submission - Geoduck Tissue & gDNA for Illumina Pilot Sequencing Project
2016
Geoduck Genome Sequencing
Samples Submitted
geoduck
Illumina
Panopea generosa
DNA Isolation - Geoduck gDNA for Potential Illumina-initiated Sequencing Project
2016
Geoduck Genome Sequencing
DNA Isolation
E.Z.N.A. Mollusc Kit
gel
geoduck
O'geneRuler DNA Ladder Mix
Panopea generosa
Qubit 3.0
Qubit dsDNA BR
Sample Submission - Geoduck Reduced Representation Bisulfite Pooled Libraries
2016
Samples Submitted
geoduck
Hollie Putnam
Panopea generosa
RRBS
Sample Submission - Ostrea lurida gDNA for PacBio Sequencing
2016
Olympia Oyster Genome Sequencing
Samples Submitted
gDNA
olympia oyster
Ostrea lurida
PacBio
Data Management - Integrity Check of Final BGI Olympia Oyster & Geoduck Data
2016
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
DNA Isolation - Ostrea lurida DNA for PacBio Sequencing
2016
Olympia Oyster Genome Sequencing
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
gDNA
gel
olympia oyster
Ostrea lurida
Qubit 3.0
Qubit dsDNA BR
Data Management - Download Final BGI Genome & Assembly Files
2016
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
BGI
geoduck
jupyter notebook
olympia oyster
Ostrea lurida
owl
Panopea generosa
Data Analysis - Continued O.lurida Fst Analysis from GBS Data
2016
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
Data Analysis - Initial O.lurida Fst Determination from GBS Data
2016
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
docker
dockerfile
FST
jupyter notebook
olympia oyster
Ostrea lurida
vcftools
Data Management - Tracking O.lurida FASTQ File Corruption
2016
Olympia Oyster Genome Sequencing
Computing - Retrieve data from Amazon EC2 Instance
2016
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
Data Management – Geoduck Small Insert Library Genome Assembly from BGI
2016
Geoduck Genome Sequencing
BGI
geoduck
jupyter notebook
Panopea generosa
Server HDD Failure – Owl
2016
Computer Servicing
Data Analysis - fastStructure Population Analysis of Oly GBS PyRAD Output
2016
Genotype-by-sequencing at BGI
fastStructure
jupyter notebook
olympia oyster
Ostrea lurida
Structure
Data Analysis - PyRad Analysis of Olympia Oyster GBS Data
2016
Genotype-by-sequencing at BGI
docker
EC2
jupyter notebook
olympia oyster
Ostrea lurida
pyrad
Computing - Not Enough Power!
2016
computing
EC2
jupyter notebook
pyrad
stacks
ustacks
Computing - Amazon EC2 Instance Out of Space?
2016
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
Data Analysis - Oly GBS Data Using Stacks 1.37
2016
Genotype-by-sequencing at BGI
BGI
GBS
genotype-by-sequencing
ipython notebook
jupyter notebook
olympia oyster
Ostrea lurida
stacks
Data Management - Olympia Oyster Small Insert Library Genome Assembly from BGI
2016
Olympia Oyster Genome Sequencing
BGI
ipython notebook
jupyter notebook
olympia oyster
Ostrea lurida
SRA Release - Transcriptomic Profiles of Adult Female & Male Gonads in Panopea generosa (Pacific geoduck)
2016
Protein expression profiles during sexual maturation in Geoduck
SRA Submissions
geoduck
Panopea generosa
PRJNA316216
SRA
SRP072283
SRX1659865
SRX1659866
Data Management - O.lurida Raw BGI GBS FASTQ Data
2016
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
Data Analysis - Subset Olympia Oyster GBS Data from BGI as Single Population Using PyRAD
2016
Genotype-by-sequencing at BGI
.vcf
IGV
ipython notebook
jupyter notebook
olympia oyster
Ostrea lurida
pyrad
SNPs
Data Management - Concatenate FASTQ files from Oly MBDseq Project
2016
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
SRA Submission – Genome sequencing of the Olympia oyster (Ostrea lurida)
2016
Olympia Oyster Genome Sequencing
SRA Submissions
NCBI
olympia oyster
Ostrea lurida
PRJNA316624
SAMN04588827
SRA
SRP072461
SRS1365663
SRX1667397
SRX1667398
SRX1667399
SRX1667400
SRX1667401
SRX1667402
SRA Submission – Genome sequencing of the Pacific geoduck (Panopea generosa)
2016
Geoduck Genome Sequencing
SRA Submissions
geoduck
NCBI
Panopea generosa
SRA
PRJNA316601
SRA Submission - Transcriptomic Profiles of Adult Female & Male Gonads in Panopea generosa (Pacific geoduck).
2016
Protein expression profiles during sexual maturation in Geoduck
geoduck
NCBI
Panopea generosa
SRA
SRP072283
Data Management - O.lurida 2bRAD Dec2015 Undetermined FASTQ files
2016
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
2bRAD
ipython notebook
jupyter notebook
olympia oyster
Ostrea lurida
RAD
RAD-seq
Data Received - Ostrea lurida MBD-enriched BS-seq
2016
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
Data Received - Bisulfite-treated Illumina Sequencing from Genewiz
2016
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Data Received - Oly 2bRAD Illumina Sequencing from Genewiz
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Sample Submission - BS-seq Library Pool to Genewiz
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Samples Submitted
Illumina Methylation Library Quantification - BS-seq Oly/C.gigas Libraries
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Illumina Methylation Library Construction - Oly/C.gigas Bisulfite-treated DNA
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Bioanalyzer - Bisulfite-treated Oly/C.gigas DNA
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Bisulfite Treatment - Oly Reciprocal Transplant DNA & C.gigas Lotterhos DNA for BS-seq
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Agarose Gel - Oly gDNA for BS-seq Libraries, Take Two
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Agarose Gel - Oly gDNA for BS-seq Libraries
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
DNA Isolation - Oly gDNA for BS-seq
2015
BS-seq Libraries for Sequencing at Genewiz
Olympia oyster reciprocal transplant
1NF11
1NF12
1NF13
1NF14
1NF15
1NF16
1NF17
1NF18
2NF1
2NF2
2NF3
2NF4
2NF5
2NF6
2NF7
2NF8
BS-seq
chloroform
DNA Isolation
DNA Quantification
DNazol
Fidalgo Bay
olympia oyster
Ostrea lurida
Oyster Bay
Qubit 3.0
Qubit dsDNA BR
RNAse A
Sample Submission - 2bRAD Libraries for Genewiz
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Samples Submitted
Sample Submission - Olympia oyster MBD-enriched DNA to ZymoResearch
2015
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
Samples Submitted
Sample Submission - Additional Olympia Oyster gDNA for Genome Sequencing @ BGI
2015
Olympia Oyster Genome Sequencing
Samples Submitted
Sample Submission - Oly Oyster Bay Tissues for GBS
2015
Genotype-by-sequencing at BGI
Olympia oyster reciprocal transplant
Samples Submitted
Samples Received - Oly Tissue & DNA from Katherine Silliman
2015
Olympia oyster reciprocal transplant
Samples Received
DNA Quality Assessment - Geoduck & Olympia Oyster gDNA
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
DNA Isolation - Olympia Oyster Outer Mantle gDNA
2015
Olympia Oyster Genome Sequencing
chloroform
DNA Isolation
DNA Quantification
DNazol
gDNA
mantle
NanoDrop1000
olympia oyster
Ostrea lurida
Qubit 3.0
Qubit dsDNA BR
DNA Isolation - Geoduck Ctenidia gDNA
2015
Geoduck Genome Sequencing
chloroform
ctenidia
DNA Isolation
DNA Quantification
DNazol
gDNA
geoduck
NanoDrop1000
Panopea generosa
Qubit 3.0
Qubit dsDNA BR
Phenol-Chloroform DNA Cleanup - Geoduck gDNA
2015
Geoduck Genome Sequencing
DNA Quantification
gDNA
geoduck
NanoDrop1000
Panopea generosa
phenol-chloroform cleanup
Quant-iT DNA BR Kit
Victor 1420 plate reader
DNA Isolation - Geoduck Adductor Muscle gDNA
2015
Geoduck Genome Sequencing
DNA Isolation
DNA Quantification
gDNA
geoduck
NanoDrop1000
Panopea generosa
Quant-iT DNA BR Kit
Victor 1420 plate reader
DNA Isolation - Olympia Oyster Outer Mantle gDNA
2015
Olympia Oyster Genome Sequencing
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
gDNA
NanoDrop1000
olympia oyster
Ostrea lurida
Quant-iT DNA BR Kit
Victor 1420 plate reader
DNA Quantification - MBD-enriched Olympia oyster DNA
2015
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
DNA Quantification
MBD
MBD-Seq
olympia oyster
Ostrea lurida
plate reader
Quant-iT DNA BR Kit
Victor 1420 plate reader
Ethanol Precipitation - Olympia oyster MBD
2015
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
EtOH precipitation
MethylMiner Methylated DNA Enrichment Kit
olympia oyster
Ostrea lurida
MBD Enrichment - Sonicated Olympia Oyster gDNA
2015
MBD Enrichment for Sequencing at ZymoResearch
Olympia oyster reciprocal transplant
hc1_2B
hc1_4B
hc2_15B
hc2_17
hc3_1
hc3_10
hc3_11
hc3_5
hc3_7
hc3_9
Hood Canal
MethylMiner Methylated DNA Enrichment Kit
olympia oyster
Ostrea lurida
Oyster Bay
ss2_14B
ss2_18B
ss2_9B
ss3_14B
ss3_15B
ss3_16B
ss3_20
ss3_3B
ss3_4B
ss5_18
DNA Sonication - Oly gDNA for MBD
2015
MBD Enrichment for Sequencing at ZymoResearch
bioanalyzer
Bioruptor 300
electropherogram
hc1_2B
hc1_4B
hc2_15B
hc2_17
hc3_1
hc3_10
hc3_11
hc3_5
hc3_7
hc3_9
Hood Canal
olympia oyster
Ostrea lurida
Oyster Bay
sonication
ss2_14B
ss2_18B
ss2_9B
ss3_14B
ss3_15B
ss3_16B
ss3_20
ss3_3B
ss3_4B
ss5_18
qPCR – Oly RAD-Seq Library Quantification
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
2bRAD
KAPA Library Illumina Quantification Kit
olympia oyster
Opticon
Opticon2
Ostrea lurida
qPCR
RAD
RAD-seq
qPCR - Oly RAD-Seq Library Quantification
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
2bRAD
KAPA Library Illumina Quantification Kit
olympia oyster
Opticon
Opticon2
Ostrea lurida
qPCR
RAD
RAD-seq
Gel Extraction - Oly RAD-Seq Prep Scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
2bRAD
gel extraction
olympia oyster
Ostrea lurida
QIAQuick Gel Extraction Kit
RAD
RAD-seq
DNA Quality Assessment - Geoduck, Oly & Oly 2SN
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
Olympia oyster reciprocal transplant
Fidalgo 2SN
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
PCR – Oly RAD-seq Prep Scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
barcodes
gel
ILL-BC1
ILL-BC10
ILL-BC2
ILL-BC3
ILL-BC4
ILL-BC5
ILL-BC6
ILL-BC7
ILL-BC8
ILL-BC9
ILL-HT1
olympia oyster
Ostrea lurida
PCR
PTC-200
Q5 High-Fidelity DNA Polymerase
RAD
RAD-seq
PCR – Oly RAD-seq Test-scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
gel
ILL-BC1
ILL-HT1
ILL-LIB1
ILL-LIB2
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
PCR
PTC-200
RAD
RAD-seq
DNA Quantification & Quality Assessment - Oly 2SN gDNA
2015
Olympia oyster reciprocal transplant
bioanalyzer
DNA Quantification
Fidalgo 2SN
gDNA
NanoDrop1000
olympia oyster
Ostrea lurida
DNA Quantification & Quality Assessment - Geoduck & Oly gDNA
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
bioanalyzer
DNA Quantification
gDNA
geoduck
NanoDrop1000
olympia oyster
Ostrea lurida
Panopea generosa
DNA Isolations – Oly Fidalgo 2SN Ctenidia
2015
Olympia oyster reciprocal transplant
DNA Isolation
E.Z.N.A. Mollusc Kit
Fidalgo 2SN
gDNA
olympia oyster
Ostrea lurida
Oyster Sampling - Oly Fidalgo 2SN, 2HL, 2NF Reciprocal Transplants Final Samplings
2015
Olympia oyster reciprocal transplant
ctenidia
E.Z.N.A. Mollusc Kit
Fidalgo 2HL
Fidalgo 2NF
Fidalgo 2SN
gill
olympia oyster
Ostrea lurida
RNAlater
DNA Isolation – Geoduck & Olympia Oyster
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
DNA Isolation
DNA Quantification
gDNA
geoduck
NanoDrop1000
olympia oyster
Ostrea lurida
Panopea generosa
Adaptor Ligation – Oly AlfI-Digested gDNA for RAD-seq
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
3ILL-NR
5ILL-NR
anti-ILL
gDNA
ligation
olympia oyster
Ostrea lurida
PTC-200
RAD-seq
T4 DNA Ligase
Restriction Digest – Oly gDNA for RAD-seq w/AlfI
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
2bRAD
AlfI
gDNA
olympia oyster
Ostrea lurida
RAD-seq
restriction digestion
Troubleshooting - Oly RAD-seq
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
RAD
RAD-seq
troubleshooting
DNA Isolations - Fidalgo 2SN Reciprocal Transplants Final Samplings
2015
Olympia oyster reciprocal transplant
ctenidia
DNA Isolation
E.Z.N.A. Mollusc Kit
Fidalgo 2SN
olympia oyster
Ostrea lurida
RNAlater
PCR - Oly RAD-seq Prep Scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
gel
ILL-BC1
ILL-BC10
ILL-BC2
ILL-BC3
ILL-BC4
ILL-BC5
ILL-BC6
ILL-BC7
ILL-BC8
ILL-BC9
ILL-HT1
ILL-LIB1
ILL-LIB2
olympia oyster
Ostrea lurida
PCR
PTC-200
Q5 High-Fidelity DNA Polymerase
RAD-seq
PCR - Oly RAD-seq Test-scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
gel
ILL-BC1
ILL-HT1
ILL-LIB1
ILL-LIB2
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
PCR
PTC-200
RAD
RAD-seq
Adaptor Ligation – Oly AlfI-Digested gDNA for RAD-seq
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
3ILL-NR
5ILL-NR
anti-ILL
ligation
olympia oyster
Ostrea lurida
PTC-200
RAD-seq
T4 DNA Ligase
Restriction Digest – Oly gDNA for RAD-seq w/AlfI
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Sample Submission - Additional Geoduck gDNA for Genome Sequencing @ BGI
2015
Geoduck Genome Sequencing
'1510071003'
gDNA
geoduck
Panopea generosa
Samples Submitted
Sample Submission - Additional Olympia Oyster gDNA for Genome Sequencing @ BGI
2015
Olympia Oyster Genome Sequencing
Samples Submitted
'1510071002'
gDNA
olympia oyster
Ostrea lurida
Agarose Gel - Geoduck & Olympia Oyster gDNA
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
Olympia oyster reciprocal transplant
gDNA
gel
geoduck
NF1A
olympia oyster
Ostrea lurida
Panopea generosa
SN49A
DNA Quantification - Pooled geoduck gDNA
2015
Geoduck Genome Sequencing
DNA Quantification
gDNA
geoduck
NanoDrop1000
Panopea generosa
gDNA Isolation - Geoduck Adductor Muscle
2015
adductor muscle
DNA Isolation
DNA Quantification
DNazol
gDNA
geoduck
NanoDrop1000
Panopea generosa
DNA Isolation - Geoduck & Olympia Oyster
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
Olympia oyster reciprocal transplant
adductor muscle
DNA Isolation
DNA Quantification
DNazol
foot
geoduck
NanoDrop1000
NF1A
olympia oyster
Ostrea lurida
Panopea generosa
RNAse A
SN49A
PCR - Oly RAD-seq Test-scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
gel
ILL-BC1
ILL-HT1
ILL-LIB1
ILL-LIB2
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
PCR
PTC-200
Q5 High-Fidelity DNA Polymerase
RAD-seq
PCR - Oly RAD-seq Test-scale PCR
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Reagent Prep
gel
ILL-BC1
ILL-HT1
ILL-HT2
ILL-LIB1
ILL-LIB2
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
PCR
PTC-200
Q5 High-Fidelity DNA Polymerase
RAD-seq
Adaptor Ligation - Oly AlfI-Digested gDNA for RAD-seq
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Reagent Prep
3ILL-NR
5ILL-NR
anti-ILL
ILL-BC1
ILL-HT1
ILL-HT2
ILL-LIB1
ILL-LIB2
ligation
olympia oyster
Ostrea lurida
PTC-200
RAD-seq
T4 DNA Ligase
Goals - October 2015
2015
Goals
geoduck
olympia oyster
Ostrea lurida
Panopea generosa
Restriction Digest - Oly gDNA for RAD-seq w/AlfI
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
Reagent Prep
AlfI
gDNA
olympia oyster
Ostrea lurida
RAD-seq
restriction digestion
Sample Submission - Olympia Oyster gDNA for Genome Sequencing @ BGI
2015
Olympia Oyster Genome Sequencing
Samples Submitted
1509191001
gDNA
olympia oyster
Ostrea lurida
Sample Submission - Geoduck gDNA for Genome Sequencing @ BGI
2015
Geoduck Genome Sequencing
1509191002
gDNA
geoduck
Panopea generosa
Samples Submitted
Uninterruptible Power Supplies (UPS)
2015
Computer Servicing
Eagle
Opticon
owl
Synology
uninterruptible power supply
UPS
Agarose Gel - Geoduck & Olympia oyster gDNA Integrity Check
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
Ethanol Precipitation - Geoduck & Olympia oyster gDNA
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
DNA Quantification
EtOH precipitation
geoduck
NanoDrop1000
olympia oyster
Ostrea lurida
Panopea generosa
Agarose Gel - Olympia oyster Whole Body gDNA Integrity Check
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
gDNA
gel
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
DNA Isolation - Olympia oyster whole body
2015
2bRAD Library Tests for Sequencing at Genewiz
Olympia oyster reciprocal transplant
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
gDNA
NanoDrop1000
olympia oyster
Ostrea lurida
Agarose Gel – Geoduck gDNA Integrity Check
2015
Geoduck Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
Panopea generosa
Genomic DNA Isolation – Geoduck Adductor Muscle & Foot
2015
Geoduck Genome Sequencing
DNA Isolation
DNA Quantification
DNazol
gDNA
geoduck
NanoDrop1000
Panopea generosa
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
Genomic DNA Isolation – Olympia oyster adductor musle & mantle
2015
Olympia Oyster Genome Sequencing
DNA Isolation
DNA Quantification
DNazol
gDNA
NanoDrop1000
olympia oyster
Ostrea lurida
Genomic DNA Isolation – Geoduck Adductor Muscle & Foot
2015
Geoduck Genome Sequencing
DNA Isolation
DNA Quantification
DNazol
gDNA
geoduck
NanoDrop1000
Panopea generosa
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
Genomic DNA Isolation - Olympia oyster adductor musle & mantle
2015
Olympia Oyster Genome Sequencing
adductor muscle
DNA Isolation
DNA Quantification
gDNA
mantle
NanoDrop1000
olympia oyster
Ostrea lurida
Genomic DNA Isolation - Geoduck Adductor Muscle & Foot
2015
Geoduck Genome Sequencing
adductor muscle
DNA Isolation
DNA Quantification
DNazol
foot
gDNA
geoduck
NanoDrop1000
Panopea generosa
Agarose Gel - Geoduck & Olympia Oyster gDNA Integrity Check
2015
Geoduck Genome Sequencing
Olympia Oyster Genome Sequencing
gDNA
gel
geoduck
O'geneRuler DNA Ladder Mix
olympia oyster
Ostrea lurida
Panopea generosa
Genomic DNA Isolation - Olympia oyster adductor musle & mantle
2015
Olympia Oyster Genome Sequencing
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
gDNA
NanoDrop1000
olympia oyster
Ostrea lurida
Genomic DNA Isolation - Geoduck Adductor Muscle & Foot
2015
Geoduck Genome Sequencing
adductor muscle
DNA Isolation
DNA Quantification
E.Z.N.A. Mollusc Kit
foot
gDNA
geoduck
NanoDrop1000
Panopea generosa
RAD-Seq Library Prep Reagents
2015
2bRAD Library Tests for Sequencing at Genewiz
Reagent Prep
AlfI
RAD-seq
Reverse Transcription – O.lurida DNased RNA 1hr post-mechanical stress
2015
Olympia oyster reciprocal transplant
cDNA
ctenidia
DNased RNA
gill
mechanical stress
olympia oyster
Ostrea lurida
reverse transcriptase
reverse transcription
qPCR - Jake's O.lurida ctenidia 1hr post-mechanical stress DNased RNA
2015
Olympia oyster reciprocal transplant
actin
ctenidia
DNased RNA
gill
mechanical stress
olympia oyster
Opticon2
Ostrea lurida
qPCR
'SR ID: 1504'
'SR ID: 1505'
SsoFast EvaGreen Supermix
RNA Quantification - O.lurida 1hr post-mechanical heat stress DNased RNA
2015
Olympia oyster reciprocal transplant
ctenidia
DNased RNA
gill
mechanical stress
NanoDrop1000
olympia oyster
Ostrea lurida
RNA quantification
Sample Submission – Olympia oyster PCRs Sanger Sequencing
2015
Olympia oyster reciprocal transplant
Samples Submitted
Server HDD Failure - Owl
2015
Computer Servicing
hard drive
HDD
owl
server
Synology
DNase Treatment - O.lurida Ctenidia 1hr Post-Mechanical Stress RNA
2015
Olympia oyster reciprocal transplant
ctenidia
DNase
gill
NanoDrop1000
olympia oyster
Ostrea lurida
RNA
RNA quantification
Turbo DNA-free
RNA Isolation – O.lurida Ctenidia 1hr Post-Mechanical Stress
2015
Olympia oyster reciprocal transplant
ctenidia
gill
mechanical stress
olympia oyster
Ostrea lurida
RNA isolation
RNAzol RT
RNA Isolation - O.lurida Ctenidia 1hr Post-Mechanical Stress
2015
Olympia oyster reciprocal transplant
ctenidia
gill
mechanical stress
olympia oyster
Ostrea lurida
RNA
RNA isolation
RNAzol RT
PCR - Sea Pen luciferase
2015
Apex Red Master Mix
gDNA
gel
luciferase
O'GeneRuler 100bp DNA ladder
PCR
Renilla reniformis
sea pen
Opticon2 Calibration
2015
calibration
FAM
Opticon
Opticon2
qPCR
troubleshooting
RNAseq Data Receipt - Geoduck Gonad RNA 100bp PE Illumina
2015
Protein expression profiles during sexual maturation in Geoduck
geoduck
Geo_pool_F
Geo_pool_M
gonad
Illumina
Panopea generosa
RNA-seq
Data Received
Sample Submission – Olympia oyster PCRs Sanger Sequencing
2015
HTGC
olympia oyster
Ostrea lurida
PCR
Sanger sequencing
Samples Submitted
Reverse Transcription - O.lurida DNased RNA Controls and 1hr Heat Shock
2015
DNased RNA
M-MLV
oligo dT
olympia oyster
Ostrea lurida
reverse transcription
Sample Submission – Olympia oyster & Sea Pen PCRs Sanger Sequencing
2015
olympia oyster
Ostrea lurida
Renilla reniformis
Sanger sequencing
sea pen
Sample Submitted
Gel Purification - Olympia Oyster and Sea Pen PCRs
2015
DNA
gel purification
olympia oyster
Ostrea lurida
PCR
Renilla reniformis
sea pen
Ultrafree-DA
Sample Submission - Olympia oyster PCRs Sanger Sequencing
2015
HTGC
olympia oyster
Ostrea lurida
PCR
Sanger sequencing
Samples Submitted
Sample Submission - Geoduck Gonad for RNA-seq
2015
Protein expression profiles during sexual maturation in Geoduck
DNased RNA
geoduck
Geo_pool_F
Geo_pool_M
gonad
HiSeq2500
Illumina
Panopea generosa
RNA-seq
Samples Submitted
Bioanalyzer - Geoduck Gonad RNA Quality Assessment
2015
Protein expression profiles during sexual maturation in Geoduck
bioanalyzer
DNased RNA
electropherogram
gel
geoduck
gonad
NanoDrop1000
Panopea generosa
Reverse Transcription - Subset of Jake’s O.lurida DNased RNA
2015
cDNA
ctenidia
DNased RNA
gill
heat shock
M-MLV
oligo dT
olympia oyster
Ostrea lurida
reverse transcriptase
reverse transcription
Bioinformatics – Trimmomatic/FASTQC on C.gigas Larvae OA NGS Data
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bioinformatics
BS-seq
Crassostrea gigas
FASTQC
Illumina
ipython notebook
jupyter notebook
NGS sequencing
Pacific oyster
Trimmomatic
qPCR - Re-run Jake's O.lurida DNased RNA Samples NC1, SC1, SC2, SC4 from 20150514
2015
actin
DNased RNA
olympia oyster
Opticon
Opticon2
Ostrea lurida
qPCR
'SR ID: 1504'
'SR ID: 1505'
SsoFast EvaGreen Supermix
qPCR – Jake's O.lurida ctenidia DNased RNA (1hr Heat Shock Samples)
2015
actin
ctenidia
DNased RNA
gill
olympia oyster
Opticon
Opticon2
Ostrea lurida
qPCR
'SR ID: 1504'
'SR ID: 1505'
SsoFast EvaGreen Supermix
qPCR – Jake's O.lurida ctenidia DNased RNA (Control Samples)
2015
actin
ctenidia
DNased RNA
gill
olympia oyster
Ostrea lurida
qPCR
'SR ID: 1504'
'SR ID: 1505'
SsoFast EvaGreen Supermix
DNase Treatment - Jake’s O.lurida Ctenidia RNA (1hr Heat Shock) from 20150506
2015
DNase
DNased RNA
NanoDrop1000
olympia oyster
Ostrea lurida
RNA
RNA quantification
Turbo DNA-free
DNase Treatment - Jake’s O.lurida Ctenidia RNA (Controls) from 20150507
2015
DNase
DNased RNA
NanoDrop1000
olympia oyster
Ostrea lurida
RNA
RNA quantification
Turbo DNA-free
ISO Creation - OpticonMonitor3 Disc Cloning
2015
Computer Servicing
Apple
disk image
ISO
Opticon
OS X
software
qPCR - Jake O.lurida ctenidia RNA (Heat Shock Samples) from 20150506
2015
actin
ctenidia
gill
heat shock
olympia oyster
Opticon2
Ostrea lurida
qPCR
RNA
'SR ID: 1504'
'SR ID: 1505'
qPCR - Jake O.lurida ctenidia RNA (Control Samples) From 20150507
2015
actin
ctenidia
gill
olympia oyster
Opticon2
Ostrea lurida
qPCR
RNA
'SR ID: 1504'
'SR ID: 1505'
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
DNased RNA
geoduck
gonad
histology cassettes
NanoDrop1000
Panopea generosa
paraffin
PAXgene Tissue RNA Kit
RNA
RNA isolation
RNA quantification
RNA Isolation – Jake’s O. lurida Ctenidia Control from 20150422
2015
ctenidia
gill
heat shock
NanoDrop1000
olympia oyster
Ostrea lurida
RNA
RNA isolation
RNA quantification
RNAzol RT
Bioinformatics - Trimmomatic/FASTQC on C.gigas Larvae OA NGS Data
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bioinformatics
BS-seq
Crassostrea gigas
FASTQC
graphs
ipython notebook
jupyter notebook
Pacific oyster
Trimmomatic
troubleshooting
RNA Isolation - Jake’s O. lurida Ctenidia 1hr Heat Stress from 20150422
2015
ctenidia
DEPC-H2O
gill
heat shock
NanoDrop1000
olympia oyster
Ostrea lurida
pics
RNA isolation
RNA quantification
RNAzol RT
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
geoduck
histology cassettes
NanoDrop1000
Panopea generosa
paraffin
PAXgene Tissue RNA Kit
RNA isolation
RNA quantification
BLAST - C.gigas Larvae OA Illumina Data Against GenBank nt DB
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bioinformatics
BLAST
BLASTn
BS-seq
Crassostrea gigas
GenBank
ipython notebook
jupyter notebook
NCBI
Pacific oyster
Goals - May 2015
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
Goals
Lineage-specific DNA methylation patterns in developing oysters
LSU C.virginica Oil Spill MBD BS Sequencing
Protein expression profiles during sexual maturation in Geoduck
BS-seq
Crassostrea gigas
Crassostrea virginica
Eastern oyster
geoduck
Pacific oyster
Panopea generosa
BLASTN - C.gigas OA Larvae to C.gigas Ensembl 1.24 BLAST DB
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BLAST
Crassostrea gigas
ipython notebook
jupyter notebook
Pacific oyster
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
geoduck
gonad
histology cassettes
Panopea generosa
paraffin
PAXgene Tissue RNA Kit
RNA isolation
RNA quantification
Bioanalyzer Data - Geoduck RNA from Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
bioanalyzer
electropherogram
foot
gel
geoduck
Panopea generosa
RNA
RNA Pico
RNA Isolation – Geoduck Gonad in Paraffin Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
DNased RNA
geoduck
gonad
histology cassettes
NanoDrop1000
Panopea generosa
PAXgene Tissue RNA Kit
RNA isolation
RNA quantification
Bioanalyzer Submission - Geoduck Gonad RNA from Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
bioanalyzer
geoduck
gonad
Panopea generosa
paraffin
RNA
Quality Trimming - C.gigas Larvae OA BS-Seq Data
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bioinformatics
BS-seq
Crassostrea gigas
FASTQC
ipython notebook
jupyter notebook
larvae
OA
ocean acidification
Pacific oyster
Trimmomatic
Quality Trimming - LSU C.virginica Oil Spill MBD BS-Seq Data
2015
LSU C.virginica Oil Spill MBD BS Sequencing
BS-seq
Crassostrea virginica
Eastern oyster
FASTQC
ipython notebook
jupyter notebook
MBD-Seq
NGS sequencing
oil
Trimmomatic
Sequence Data Analysis - LSU C.virginica Oil Spill MBD BS-Seq Data
2015
LSU C.virginica Oil Spill MBD BS Sequencing
BS-seq
Crassostrea virginica
Eastern oyster
FASTQC
LSU
MBD-Seq
NGS sequencing
oil
Sequence Data Analysis - C.gigas Larvae OA BS-Seq Data
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BS-seq
Crassostrea gigas
FASTQC
larvae
NGS sequencing
OA
ocean acidification
Pacific oyster
Sequence Data - C.gigas OA Larvae BS-Seq Demultiplexed
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BS-seq
Crassostrea gigas
larvae
NGS sequencing
OA
ocean acidification
Pacific oyster
Sequence Data - LSU C.virginica Oil Spill MBD BS-Seq Demultiplexed
2015
LSU C.virginica Oil Spill MBD BS Sequencing
BS-seq
Crassostrea virginica
Eastern oyster
LSU
MBD-Seq
NGS sequencing
oil
RNA Isolation - Geoduck Gonad in Paraffin Histology Blocks
2015
Protein expression profiles during sexual maturation in Geoduck
Disruptor Genie
geoduck
gonad
histology
histology cassettes
NanoDrop1000
Panopea generosa
paraffin
PAXgene Tissue RNA Kit
RNA isolation
RNA quantification
Sequencing Data - C.gigas Larvae OA
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BS-seq
checksum
code
Crassostrea gigas
curl
HiSeq2500
larvae
md5
OA
ocean acidification
Pacific oyster
TruSeq Adaptor Counts – LSU C.virginica Oil Spill Sequences
2015
LSU C.virginica Oil Spill MBD BS Sequencing
TruSeq Adaptor Identification Method Comparison - LSU C.virginica Oil Spill Sequences
2015
LSU C.virginica Oil Spill MBD BS Sequencing
barcodes
Crassostrea virginica
Eastern oyster
FASTQ
FASTQC
fastx_barcode_splitter
grep
index sequences
ipython notebook
jupyter notebook
TruSeq
DNA Quantification - Claire’s C.gigas Sheared DNA
2015
Lineage-specific DNA methylation patterns in developing oysters
Crassostrea gigas
DNA Quantification
heat shock
Pacific oyster
Quant-iT DNA BR Kit
Library Quality Assessment - C.gigas OA larvae Illumina libraries
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bioanalyzer
BS-seq
Crassostrea gigas
EpiNext Post-Bisulfite DNA Library Preparation Kit
Genomics Core Facility
larvae
library prep
OA
ocean acidification
Pacific oyster
University of Oregon
BS-seq Library Prep - C.gigas Larvae OA 1000ppm
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bisulfite conversion
BS-seq
Crassostrea gigas
EpiNext Post-Bisulfite DNA Library Preparation Kit
larvae
library prep
Methylamp DNA Modification
NanoDrop1000
OA
ocean acidification
Pacific oyster
DNA Quantification - C.gigas Larvae 1000ppm
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
Crassostrea gigas
DNA
larvae
NanoDrop1000
OA
ocean acidification
Pacific oyster
DNA Quantification - Claire’s Sheared C.gigas Mantle Heat Shock Samples
2015
Lineage-specific DNA methylation patterns in developing oysters
Crassostrea gigas
DNA Quantification
graphs
heat shock
mantle
NanoDrop1000
Pacific oyster
Bioanalyzer - C.gigas Sheared DNA from 20140108
2015
Lineage-specific DNA methylation patterns in developing oysters
bioanalyzer
Crassostrea gigas
DNA
DNA1000
electropherogram
heat shock
mantle
Pacific oyster
spreadsheet
Library Prep - Quantification of C.gigas larvae OA 1000ppm library
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BS-seq
Crassostrea gigas
DNA Quantification
FLx800 plate reader
larvae
library prep
OA
ocean acidification
Pacific oyster
Quant-iT DNA BR Kit
Sequencing Data - LSU C.virginica MBD BS-Seq
2015
LSU C.virginica Oil Spill MBD BS Sequencing
BS-seq
checksum
code
Crassostrea virginica
curl
Eastern oyster
HiSeq2500
LSU
MBD
md5
oil
Data Received
Bisulfite NGS Library Prep - Bisulfite Conversion & Illumina Library Construction of C.gigas larvae DNA
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bisulfite conversion
BS-seq
Crassostrea gigas
DNA
larvae
library prep
OA
ocean acidification
Pacific oyster
Bisuflite NGS Library Prep – C.gigas larvae OA bisulfite library quantification
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
BS-seq
Crassostrea gigas
DNA Quantification
FLx800 plate reader
library prep
OA
ocean acidification
Pacific oyster
Quant-iT DNA BR Kit
Bisuflite NGS Library Prep - C.gigas larvae OA bisulfite DNA (continued from yesterday)
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
barcodes
BS-seq
Crassostrea gigas
library prep
Pacific oyster
Bisuflite NGS Library Prep - C.gigas larvae OA bisulfite DNA
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
Crassostrea gigas
larvae
library prep
Methylamp DNA Modification
OA
ocean acidification
Pacific oyster
Library Cleanup - LSU C.virginica MBD BS Library
2015
LSU C.virginica Oil Spill MBD BS Sequencing
bioanalyzer
Crassostrea virginica
DNA Quantification
Eastern oyster
electropherogram
library prep
DNA Bisulfite Conversion - C.gigas larvae OA Sheared DNA
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
bisulfite conversion
DNA
SpeedVac - C.gigas larvae OA DNA
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
concentrate
Crassostrea gigas
DNA
SpeedVac
DNA Isolation - C.gigas larvae from 2011 NOAA OA Experiment
2015
Crassostrea gigas larvae OA (2011) bisulfite sequencing
Crassostrea gigas
DNA Isolation
DNA Quantification
DNA Shearing
larvae
OA
ocean acidification
Bisulfite NGS Library Prep - LSU C.virginica Oil Spill MBD Bisulfite DNA and Emma’s C.gigas Larvae OA Bisulfite DNA (continued from yesterday)
2014
Crassostrea gigas larvae OA (2011) bisulfite sequencing
LSU C.virginica Oil Spill MBD BS Sequencing
Bisulfite NGS Library Prep - LSU C.virginica Oil Spill Bisulfite DNA and Emma’s C.gigas Larvae OA Bisulfite DNA
2014
Crassostrea gigas larvae OA (2011) bisulfite sequencing
LSU C.virginica Oil Spill MBD BS Sequencing
Bisulfite Conversion - LSU C.virginica Oil Spill MBD DNA and Emma’s C.gigas Larvae OA DNA
2014
Crassostrea gigas larvae OA (2011) bisulfite sequencing
LSU C.virginica Oil Spill MBD BS Sequencing
DNA Isolation - Claire’s C.gigas Female Gonad for Illumina Bisulfite Sequencing
2014
Lineage-specific DNA methylation patterns in developing oysters
Gel - Sheared gDNA
2014
LSU C.virginica Oil Spill MBD BS Sequencing
DNA Isolation - C.gigas Larvae from Emma OA Experiments
2014
Crassostrea gigas larvae OA (2011) bisulfite sequencing
RAD Sequencing - Oly Oyster gDNA-01 RAD Library (from 20141110)
2014
Olympia oyster reciprocal transplant
Samples Submitted
library prep
olympia oyster
Ostrea lurida
RAD-seq
Library Prep - Oly Oyster gDNA-01 RAD
2014
Olympia oyster reciprocal transplant
Ligation - Illumina P1 Adapters for Oly Oyster gDNA-01 RAD Sequencing (from 20141031)
2014
Olympia oyster reciprocal transplant
Restriction Digest - Oly Oyster gDNA-01 for RAD Sequencing (from 20141029)
2014
Olympia oyster reciprocal transplant
DNA Isolation - Claire’s C.gigas Female Gonad
2014
Lineage-specific DNA methylation patterns in developing oysters
DNA Isolation - Test Sample
2014
Lineage-specific DNA methylation patterns in developing oysters
Miscellaneous
Phenol-Chloroform DNA Clean Up - Mac and Claire’s Samples (from 20140410)
2014
Lineage-specific DNA methylation patterns in developing oysters
DNA Isolation - Claire’s C.gigas Female Gonad and Mac’s C.gigas Gonad
2014
Lineage-specific DNA methylation patterns in developing oysters
DNA gel - Claire’s C.gigas Female Gonad and Mac’s C.gigas Gonad
2014
Lineage-specific DNA methylation patterns in developing oysters
DNA Isolation - C.gigas Female Gonads (from frozen)
2014
Lineage-specific DNA methylation patterns in developing oysters
Illumina RNAseq Library Construction - 32 C.gigas Individuals
2012
Crassostrea gigas
Illumina
library prep
Pacific oyster
RNA-seq
Sample Submission - QPX RNA and DNA for Illumina 36bp single-end RNA/DNAseq
2012
Miscellaneous
Samples Submitted
Reverse Transcription - DNased C.gigas Larval RNA from 20120427
2012
Miscellaneous
M-MLV
reverse transcription
RNA
Vibrio tubiashii
qPCR - Check DNased RNA from Earlier Today for Residual gDNA
2012
CFX96
Crassostrea gigas
DNased RNA
larvae
Pacific oyster
qPCR
Vibrio tubiashii
VtpA
qPCR - C.gigas V.vulnificus Exposure cDNA (from 20110311)
2011
cDNA
CFX96
Crassostrea gigas
defensin
GAPDH
Pacific oyster
qPCR
Vibrio exposure
Vibrio vulnificus
Primer Design - Hard Clam NGS Primers
2011
Miscellaneous
Hard clam
Mercenaria mercenaria
primers
qPCR - Hard Clam Primers on cDNA from yesterday
2010
Miscellaneous
BigDef
C1qTNG
CA
CFX96
CytP450-like
ferritnin
graphs
GST
Hard clam
HemoDef
HSP 70
Immomix
lysozyme
MA
MAX
Mercenaria mercenaria
Mercenaria_Rel
metallothionein
Mm_TRAF6
NADHIV
qPCR
RACK1
SenescentProt
STI
SYTO 13
TfAP1
TLR
gDNA Isolation - Mac gigas larvae samples: control larvae 6.7.10 and 5-aza tr larvae 6.7.10
2010
Miscellaneous
5-azacitidine
Crassostrea gigas
DNA Isolation
DNA Quantification
DNazol
gDNA
larvae
NanoDrop1000
Pacific oyster
gDNA Isolation - Mac gigas gill samples (continued from yesterday)
2010
Miscellaneous
Crassostrea gigas
DNA Isolation
DNA Quantification
DNazol
gDNA
gel
gill
NanoDrop1000
Pacific oyster
R37
R51
RNAlater
gDNA Isolation - Mac gigas gill samples
2010
Miscellaneous
Crassostrea gigas
DNA Isolation
DNazol
gill
Pacific oyster
R37
R51
RNAlater
Package Rec’d - From NOAA in Connecticut
2010
Miscellaneous
algae
Isochrysis sp. T-150
NOAA
package
Tetraselmis cheri Ply429
Thalassiaosira weissflugii TW
gDNA Isolation - Mac gigas gill samples
2010
Miscellaneous
Crassostrea gigas
DNazol
Pacific oyster
R51
RNAlater
DNA Isolation - Qiagen Kit Comparison
2010
Miscellaneous
DNeasy
gDNA
gel
Hyperladder I
troubleshooting
Reverse Transcription - cDNA from DNased Abalone RNA from 20090420
2009
Miscellaneous
black abalone
DNased RNA
Haliotis cracherodii
M-MLV
reverse transcription
RNA
PCR - Bay/Sea scallop gDNA isolated earlier today
2009
Miscellaneous
RNA - Hard clam hemo RNA (from 20090121)
2009
Miscellaneous
Hard clam
hemocyte
Mercenaria mercenaria
mRNA
mRNA enrichment
SDS/PAGE, Western Blot - Test of new Western Breeze Kit & HSP70 Ab for FISH441
2008
Miscellaneous
anti-HSP70
anti-myc
antibody
Coomassie
Crassostrea gigas
MSTN
MSTN1b
myostatin
Pacific oyster
protein
SDS-PAGE
SeeBlue Plus
Western blot
Western Breeze Chromogenic (anti-mouse) Kit
RNA Isolation - V. tubiashii from challenge (see 20081216)
2008
Miscellaneous
MICROBExpress Kit
NanoDrop1000
RNA isolation
RNA quantification
TriReagent
Vibrio tubiashii
Vibrio challenge
2008
Miscellaneous
bacterial challenge
bacterial culture
Vibrio exposure
Vibrio tubiashii
SDS/PAGE/Western - anti-HSP70 Ab Re-test
2008
Miscellaneous
anti-HSP70
antibody
chemiluminescent
Coomassie
Crassostrea gigas
hemocyte
hemolymph
Pacific oyster
protein
SDS-PAGE
SeeBlue Plus
SDS/PAGE/Western - anti-HSP70 Ab test
2008
Miscellaneous
anti-HSP70
antibody
Coomassie
Crassostrea gigas
gill
mucus
Octopus rubescans
Pacific oyster
protein
red octopus
SDS-PAGE
skin
Sequencing - Y2H colony PCRs from 20081112
2008
Myostatin Interacting Proteins
No matching items